Синтез пиррольных интермедиатов для высокосопряженных порфиринов
Исторически большинство исследований в области химии порфиринов были направлены на изучение, синтез и определение биохимических свойств природных тетрапиррольных пигментов (гемов, хлорофиллов, и т.д.), но в последние 20 лет наблюдается перенаправление усилий для синтеза нестандартных порфириновых систем и использования их в новых материалах. Например, порфиринсодержащие системы исследуются на возможность применения в качестве молекулярных механизмов, молекулярных проводников и молекулярных фотонных проводников. Порфирины и структурно родственные им фталоцианины всесторонне исследуются на возможность их применения в жидкокристаллических мониторах и светопоглощающих материалах.
Большинство исследований порфиринов ранее проводилось с использованием мезо-тетраарилпорфиринов, частично из-за того, что эти системы могут быть легко получены по известным литературным методам. Модифицированные системы, которые имеют хромофорные системы, поглощающие в красной и инфракрасной областях спектра, имеют огромное значение и это стимулирует дальнейшие исследования порфириновых аналогов, включающих фуран- и тиофенсодержащие макроциклы, структурные изомеры порфиринов, и так называемые расширенные порфирины. Порфирины, расширенные ароматическими фрагментами, могут выступать в качестве высоко модифицированных хромофоров, при этом сохраняя свойства, связанные с порфириновым ядром. (1)
Расширение p‑электронной порфиринового макроцикла при введение бензольного кольца приводит к значительному сдвигу в красную область спектра и уменьшает окислительный потенциал этих соединений. Высоко сопряженные порфирины, например бензопорфирины (1) и пиридинопорфирины (2), проявляют некоторые свойства, которые могут быть использованы в области медицины и новых материалов для электронной техники. (2,3).
Новым этапом в химии высоко сопряженных порфиринов явилась дальнейшая модификация хромофорной системы порфиринового ядра путем введения более сложных ароматических и гетероароматических систем. На этом этапе наряду с традиционным методом расширения порфиринового макроцикла – наращиванием ароматического ядра на основе функционально замещенного порфирина (2,3,4), был разработан новый более продуктивный метод, заключающийся в формировании сопряженной по b-положениям пиррольного кольца системы пиррола и ароматического фрагмента, с последующим введением этой системы в порфириновую конденсацию (5,6,7). На основе последнего подхода были синтезированы фенантропорфирины (3) и фенантролинопорфирины (4), а также тиадиазолопорфирины (5), которые нашли применение в качестве молекулярных зондов, фотосенсоров в фотодинамической терапии рака, геохимических стандартов при анализе осадочных металлопорфиринов (7).
Данная работа посвящена синтезу пиррольных интермедиатов для высоко сопряженных порфиринов, которые являются ключевым звеном в реализации последнего подхода.
2. Литературный обзор.
2.1. Синтез замещенных пирролов.
Для синтеза пиррольного цикла предложено большое количество методов. В этой работе будут рассмотрены лишь некоторые из них.
2.1.1. Образование связей C–N и С–С в результате реакции аминогруппы и метиленовой группы с карбонильной.
Синтез Кнорра наиболее общий и широко используемый метод получения пирролов (8). Он заключается в конденсации a-аминокетонов и a-амино-b-кетоэфиров с кетонами или кетоэфирами в присутствии уксусной кислоты и реже щелочи. Реакции обычно протекают с хорошим выходом. a-Аминокетоны получают восстановлением цинком в уксусной кислоте из предварительно полученных изонитрозо-b-кетоэфиров или изонитрозо-b‑дикетонов(9,10).
Модификацией этого метода является конденсация предварительно приготовленной соли аминокетона с кетоэфиром в щелочной среде (11). При этом получаются пирролы, содержащие в положениях 2 и 3 метильные группы, а в положениях 4 и 5 электроноакцепторные заместители. Кроме указанного восстановителя успешно был применен дитионит натрия (12).
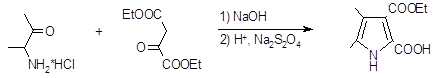
В качестве изонитрозокетонов и изонитрозокетоэфиров могут быть использованы нитрозомалоновый или нитрозоциануксусный эфиры, которые после восстановления в условиях конденсации Кнорра реагируют с b-дикетонами (13).

Синтез пирролов по Кнорру удобнее проводить без выделения промежуточного a‑аминокетона (14). Исследование этой реакции показало, что основным ограничением при использовании этой реакции является склонность a-аминокетонов к димеризации (15).
Механизм реакции Кнорра можно представить следующим образом:
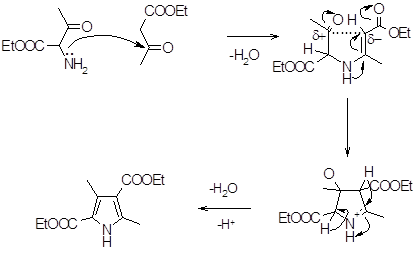
По этому же механизму протекает синтез пирролов по Кнорру в модификации Трейбса, при этом сначала происходит восстановление гидразона a-дикетона до гидразина, а затем образование енамина и альдольная конденсация протекающая с хорошими выходами 60 ‑ 70% (16).
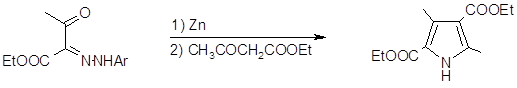
Было показано, что при конденсации 3-(b-диэтиламино)этил-пентан-2,4-диона (3) изонитрозоацетоуксусным эфиром (1)* наряду с 2,4‑диметил‑3‑(b‑диэтиламино)этил-5‑карбоэтокси-пирролом (8) образуется 3‑ацетилпиррол (7) (17,18). Если вместо ацетоуксусного эфира использовать малоновый эфир (2), то образуется 2,4-диметил-5-карбоэтоксипиррол (9).

Реакция может протекать либо по пути А с участием ацильной группы; либо по пути Б, когда ацетильная группа участвует в построении гетероцикла. В случае циклизации пентандиона (3) с b-диэтиламиноэтильной группой с изонитрозоацетоуксусным эфиром реакция осуществляется по механизму А. Конденсация пентандиона (3) с изинитрозодиэтилмалоновым эфиром происходит по пути Б.
Ещё одним классом соединений, широко используемых в синтезе пирролов, являются нитроалкены (19). Так при конденсации b-метил-b-нитростирена (10) с метилацетоацетатом (11) образуется промежуточное соединение (12). При конденсации этого же стирена с этилацетоацетатом (13) образуется другое промежуточное соединение (14), однако оба этих промежуточных соединения дают пирролы одного строения.
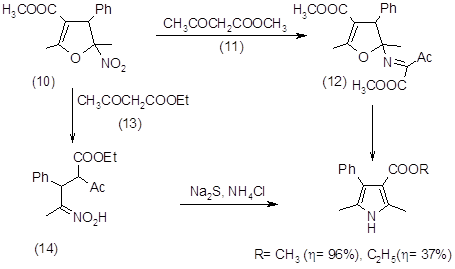
В последние годы, в связи с необходимостью введения в порфириновый макроцикл сложных ароматических систем, таких как фенантрен или фенантролин , нафталин и аценафталин, бензол (7,20,21), а также ацетали нитроацетальдегида (22) был разработан синтез пирролов, конденсированных по b‑положениям с выше указанными ароматическими системами на основе метода Бартона‑Зарда, которые показали, что нитроалкены способны при конденсации с изоцианоацетатами в присутствии ненуклеофильного основания давать пиррол-2-карбоксилаты. Хотя сам нитробензол не реагирует таким образом, другие нитроарены с большим числом двойных связей конденсируются с изоцианоацетатами, давая соответствующие пирролы с хорошим выходом до 50%.

Также были предприняты попытки синтеза пирролов, конденсированных с гетероароматическими и гетероциклическими системами, однако нитротиофен, нитрофуран и нитропиридин дали в качестве продуктов смолу, хотя 4-нитро-2,1,3-бензотиадиазол при конденсации с изоцианоацетатом дает в качестве одного из продуктов соответствующий пиррол (21).
2.1.2. Конденсации, при которых в готовый углеродный скелет вводится атом азота при помощи аммиака или аминов.
Наиболее важной в этой группе реакций является взаимодействие 1,4-дикарбонильных соединений с аммиаком по Паалю-Кнорру (19,23). Механизм реакции, очевидно, включает нуклеофильное присоединение аммиака к двум карбонильным атомам углерода и последующее отщепление двух молекул воды (24).
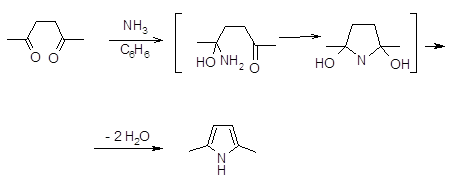
Отмечено также, что реакция может протекать с ацетатом аммония c хорошим выходом (~ 70%), причем, чем более электроноакцепторные заместители в 1,4-дикетоне, тем в более жестких условиях протекает реакция (25).
К этой же группе реакций можно отнести получение пирролов по Ганчу из a‑галогенкетонов, b-кетоэфиров и аммиака (26).
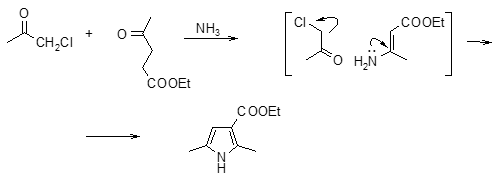
Согласно предложенному механизму, сначала происходит образование С-С связи и возникает g-дикетон, который далее реагирует с амином. К реакциям этой группы относится также взаимодействие аммиака и аминов с полиокси- и полигалоидными соединениями (27).
1H-пирролы также образуются в результате термической 1,5-сигматропной перегруппировки 3Н-пирролов, которые образуются по реакции Пааля-Кнорра (28).

Однако представленный выше метод не отличается хорошей селективностью, в результате реакции образуются изомеры, которые дают множество побочных продуктов при дальнейшей обработке. Низкий выход целевого продукта (~ 20%), а также общее время протекания всех стадий (от 90 до 160 часов) делают этот метод малоприемлемым для широкого применения.
Метод получения 2,5-диметилпиррола (17) из 2,5-гександиона (15) и гексаметилдисилазана (ГМДС) в присутствии трифторметансульфокислоты является отличным по выходу-100%, но дорогим из-за использования трифторметансульфокислоты. Реакция протекает через промежуточное соединение (16). Попытки получить 2,5-диметилпиррол (17) из 2,5‑диметилфурана (18) через это соединение закончились неудачно (29).
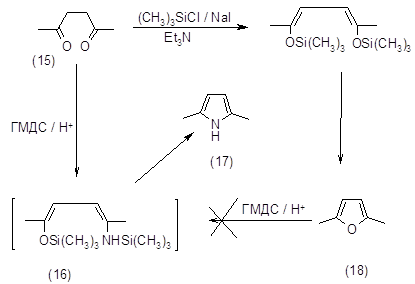
2.1.3. Реакции присоединения аминов по кратным связям.
При реакции эфиров ацетиленкарбоновой кислоты с различными нуклеофилами образуются производные пиррола (30). Первая стадия заключается в присоединении по Михаэлю, образующееся при этом соединение циклизуется затем по ниже приведенному механизму.

К этой же группе реакций можно отнести образование производных пиррола из N‑замещенных эфиров b-аминокротоновой кислоты (19) и 1-нитропропена (20), которые в реакции типа Михаэля дают аддукт, превращающийся в пирролин, окисляющийся затем в пиррол (21) (31,32).
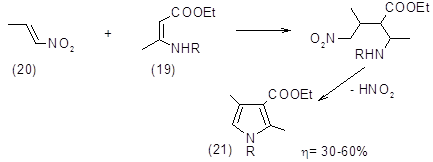
Производные пиррола из эфиров N-тозилглицина и a,b-ненасыщенных кетонов являются промежуточными в синтезе порфиринов (33).

Присоединение по Михаэлю эфира N-тозилглицина (23) к кетону (22) в присутствии третбутилата калия с последующей внутримолекулярной альдольной конденсацией приводит к образованию гидроксипирролидина (24), при дегидрогалогенировании которого образуется Δ3-пирролин (25); отщепление от него п-толуолсульфиновой кислоты под действием этилата натрия приводит к пирролу (26) с удовлетворительным выходом ~ 30-40% (16).
Ещё одним способом получения пирролов является получение пирролов по Хьюгенсу с помощью 1,3-диполярного присоединения диэфиров ацетилендикарбоновых кислот к азолактонам, образующимся в результате циклизации N-замещенных фенилглицинов в присутствии уксусного ангидрида (16). В результате таутомерии азолактон находится в равновесии с ароматическим азометинилидом, к которому присоединяется диэфир ацетилендикарбоновой кислоты с образованием бициклического интермедиата, при отщеплении от которого CO2 образуется тетразамещенный пиррол с выходом 70-80% (34).
.
Синтез замещенных пирролов может быть также осуществлен с хорошим выходом реакцией γ-кетоалкинов с бензиламином или аммиаком. Механизм реакции, вероятно, включает образование имина, который подвергается 5-экзодициклизации с последующей изомеризацией давая пиррол (35).

В последние годы было обнаружено, что при взаимодействии 1,4-дикарбонильных соединений с солями аммония по Паалю-Кнорру, могут образовываться как производные пиррола, так и производные фурана (25, 36).
.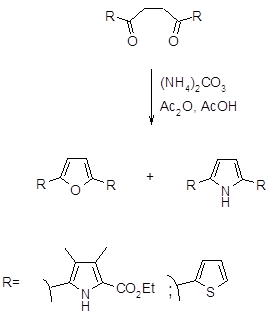
2.1.4. Реакции образования циклов в результате внутримолекулярной конденсации.
Одним из важных ограничений синтеза пирролов по Кнорру является склонность a‑аминокетонов к автоконденсации. Поэтому был разработан метод синтеза пирролов из a‑аминоамидов Вейнреба, которые проявляют низкую склонность к автоконденсации и возможность превращения в кетоны, которые затем циклизуются давая соответствующие пирролы.
Ключевой стадией процесса является превращение N‑метокси‑N‑метил‑a‑енамино-карбоамидов (27) в карбонильные производные (28) под действием металлорганических соединений. На этой стадии металлорганическое соединение (МеLi, MeMgI) селективно атакует амидную группу. При этом сначала под действием металорганического соединения отщепляется протон, давая сопряженный анион (30) к которому присоединяется металлорганическое соединение, давая интермедиат (31) при гидролизе которого образуется карбонильное соединение (28), причем здесь же может происходить частичная или полная циклизация до пиррола (29). Дальнейшая циклизация карбонильного соединения (28) до пиррола (29) происходит в условиях щелочного катализа (NaOC2H5/ C2H5OH), выходы пирролов составляют 60-70% в зависимости от заместителей (37, 38).

В данном случае из-за природы R1, R2 и Z происходит циклизация по Кнорру, однако если Z= ацил, то циклизация может происходить как по Кнорру (путь А), так и по Фишеру-Финку (путь Б) (18).

Ещё одним способом получения пирролов является йодоциклизация алкенилзамещенных b-енаминоэфиров и кетонов с последующим дегидрогалогенированием. Основой для реакции циклизации является имин-енаминное таутомерное равновесие, в котором преобладает енамин.
Было показано, что продукт электрофильной циклизации зависит от относительного положения алкенильной цепи в енаминокетоне, так из a-алкенил-bаминокетонов образуется дигидропирролы (32), а из γ-алкенил-b-аминокетонов- 2-метиленпирролидины (33) (39).

Также было отмечено, что скорость йодоциклизации зависит от заместителя у атома азота, в случае наличия сопряжения скорость увеличивается в 10-20 раз (40).
Дегидрогалогенирование дигидропиррола производят с помощью основания или без него, причем скорость дегидрогалогенирования подчиняется тем же закономерностям, что и скорость йодоциклизации.

Широко используемым может быть метод получения тетразамещенных пирролов из ненасыщенных тиоамидов, включающий образование изотиазолевых солей в качестве интермедиатов в синтезе тиазинов, при удалении из которых серы образуются тетразамещенные пирролы (23).
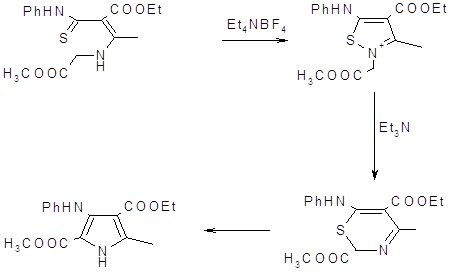
Из- за сложностей возникающих при попытках получить аминопирролы из незамещенных пирролов, был разработан метод синтеза 3-аминопирролов с хорошими выходами при помощи циклизации Торпа‑Циглера. Метод основан на N-алкилировании b-енаминонитрилов a- галогенкетонами с последующей внутримолекулярной конденсацией по Торпу‑Циглеру (41,42).
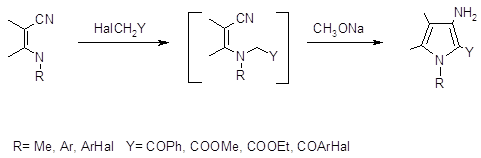
Основным требованием данной реакции является выбор алкилирующих агентов имеющих активное метиленовое звено, необходимое для протекания последующей внутримолекулярной конденсации, которая протекает спонтанно или в условиях щелочного катализа метилатом натрия.
2.2. Методы синтеза порфиринов.
Характерной чертой современной химии порфиринов является наличие значительного числа разнообразных методов построения порфиринового макроцикла. Наряду с методами, предложенными в последние годы, достаточно широко используются способы, разработанные в 20-30 годы. Новые методы, потеснив старые, полностью их не исключили. Такое, в общем, не характерное для иных классов биологически-активных соединений, состояние связано, главным образом, с тем, что синтез порфиринов зависит от характера имеющихся в порфирине заместителей и их расположения в макроцикле.
Методы, позволяющие получать сравнительно простые порфирины, совершенно не применимы к более сложным природным порфиринам. И напротив, общие методы, предложенные в последние годы, нецелесообразно применять для получения ряда порфиринов, включая и некоторые природные, поскольку в этом случае неоправданно усложняется синтез исходных пирролов (43).
Синтез порфиринов можно осуществлять двумя основными методами:
1) синтез функциональных порфиринов из непорфириновых предшественников;
2) синтез функциональных порфиринов из заранее полученных незамещенных порфиринов.
Сначала рассмотрим первый, наиболее распространенный, метод получения порфиринов.
2.2.1. Метод Адлера-Лонго.
Метод Адлера-Лонго разрабатывался в середине ХХ века, его часто используют для получения несимметричных замещенных тетраарилпорфиринов. Главной особенностью метода является получение полностью мезо - замещенного порфирина, в мезо‑ положение можно ввести как ароматические заместители (44,45), так и алифатические (46).
Суть метода заключается в конденсации α–незамещенного пиррола с альдегидом, при этом в конденсацию можно вводить смесь различных пирролов и альдегидов, но при этом образуется смесь из всех возможных комбинаций порфиринов и расширенных порфириновых циклов, содержащих 5 и более пиррольных фрагментов (47,48).
Монопиррольная тетрамеризация широко используется, когда целевым является высокосимметричный порфирин, например октаэтилпорфирин или тетрафенилпорфирин, при этом в конденсацию вводят пирролы, у которых в одном из a-положений находится метиленовая группа с легко уходящим остатком, а другое – свободно (49,50).
В последние годы были разработаны методы получения несимметричных порфиринов данным способом, но они требуют тщательного хроматографического разделения продуктов реакции (51).
Тем не менее, оказалось возможным предсказать образование требуемого порфирина, если использовать пиррол, содержащий во втором положении такую уходящую группу, которая была бы настолько реакционноспособна, чтобы ее можно было заместить без кислотного катализа, а образующийся порфириноген быстро бы окислялся в порфирин. Идеальной уходящей группой оказалась диметиламинометильная группа, благодаря которой конденсацию можно проводить действительно в нейтральных условиях. Более того, оказалось возможным получить региохимически чистый порфирин (с одинаковыми пиррольными кольцами напротив друг друга) при использовании пиррола с двумя диметиламинометильными группами и пиррола, не содержащего заместителей в a-положениях пиррольного цикла, выход составил 10-20% (52).
При необходимости реакционную способность диалкиламинометильной группы увеличивают путем проведения реакции кватернизации, при этом выход порфириновой конденсации увеличивается до 20%.
2.2.2. “2+2” порфириновый синтез.
Этот метод основан на конденсации двух молекул дипиррометанов. Существенным преимуществом синтезов порфиринов через дипирролилметаны является то, что последние получаются с достаточно высокими выходами, особенно при наличии электронодонорных заместителей в пиррольном цикле (53).
Первоначальный метод Макдональда включал использование одного дипиррометана, содержащего две формильные группы в α‑положениях и другого α‑свободного или содержащего карбоксильные группы дипиррометана (33,54,55).
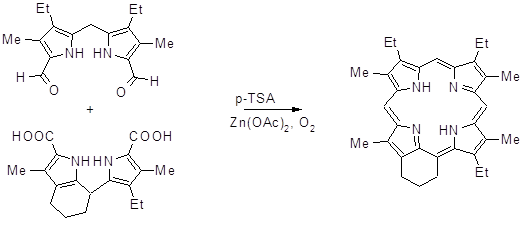
Линдсей с соавторами синтезировали порфирины, содержащие 4 различных мезозаместителя, с помощью модифицированного метода Макдональда (56,57). В качестве заместителей в мезо- положениях выступали алкил- и галоген замещенные бензолы, выход требуемого продукта достаточно высок для порфиринового синтеза и составил 14%.
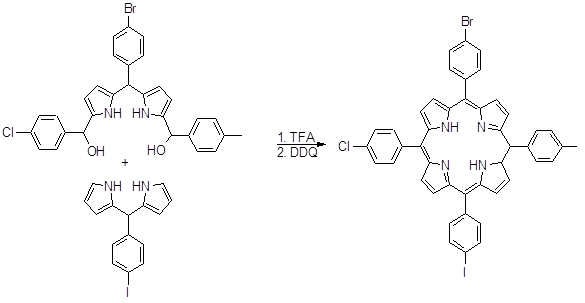
Оно и сотрудники использовали 5,5’-бис(гидроксиметил)дипиррометаны для получения несимметричных порфириновых димеров (58). Андерсон синтезировал порфириновые димеры, используя замещенные дипиррометан и триметилсилилпропиналь, как один из углеродных фрагментов. Несимметричные мезо-арилпорфирины с заместителями в β‑положениях пирольного цикла также были синтезированы данным способом. Огоши с соавторами синтезировали 5,15-дифенил-10-р-хлорфенил-2,3,17,18-тетраэтилпорфирин с выходом 9% (59).
Другой модификацией этого метода является синтез Смита, заключающийся в конденсации α‑свободных дипиррометанов и ароматического альдегида (60,61). Возможна конденсация двух молекул пиррометана, содержащего в одном из α‑положений карбонильную группу, при этом получаются зеркальносимметричные порфирины. В последние годы в качестве углеродного фрагмента, соединяющего две молекулы пиррометана, используют не только ароматические альдегиды, но и, например известны синтезы, в которых в конденсацию вводят карбальдегиды ферроцена, выход этих реакций достигает 50% (62). Порфирины, полученные этим способом, например, мезо-тетракис(1,3-диметилимидазол-2-ил)порфирин и мезо-тетракис(1,2-диметилпиразол-4-ил)порфирин, способны различным путем встраиваться в молекулу ДНК и могут быть использованы в качестве молекулярного транспорта (63).

Смит и сотрудники синтезировали мезоарил-замещенные порфирины, состоящие из двух зеркально‑симметричных фрагментов. Авторами были получены тетраарилпорфирины с различными арильными заместителями по ниже приведенной схеме. 5,15-Ди(4-толил)-10,20-дифенилпорфирин получают данным способом с 31% выходом, а 5,15-бис(4-фторо-3-метилфенил)-10,20-(4-метоксифенил)порфирин – с выходом 24% (60,64).

В дипирролилметане (34), имеющем две близкие по реакционной способности группы в положениях 5 и 5’, проводят активацию формильной группы, превращая ее через основание Шиффа (35) в тиоальдегид (36). Конденсация соединений (36) и (37), проводимая в мягких условиях, приводит лишь к одному соединению (38), замыкание и перегруппировка последнего приводят к хлорину (39) с высоким выходом.
2.2.2.1 Синтез порфиринов через b-билены.
В настоящее время известно значительное число методов получения как частично симметричных, так и полностью несимметричных порфиринов через b-билены.
Этот метод достаточно широко используется для синтеза сложных порфиринов, этому способствует ряд обстоятельств: а) более легкий синтез исходных дипирролилметанов по сравнению с получением аналогичных 5,5’-диалкоксикарбонильных соединений, которые необходимы в методе Макдональда; б) отсутствие гексапирродиенов при синтезе биленов; в) возможность получения полностью несимметричных порфиринов. Следует отметить, что при синтезе диацетилпорфирина было показано, что при получении порфиринов с одной и особенно с двумя электроотрицательными группами синтез биленов надо планировать таким образом, чтобы эти заместители находились в крайних пиррольных кольцах. При наличии подобных заместителей в дипирролилметановом фрагменте билена повышалась чувствительность метинового мостика к электрофильным реакциям. Возможность побочных реакций особенно возрастала, когда два таких заместителя расположены в противоположных пиррольных кольцах. С учетом сказанного был синтезирован замещенный цитопорфирин (40), который является ключевым соединением при получении порфирина α, выход составил 30%.
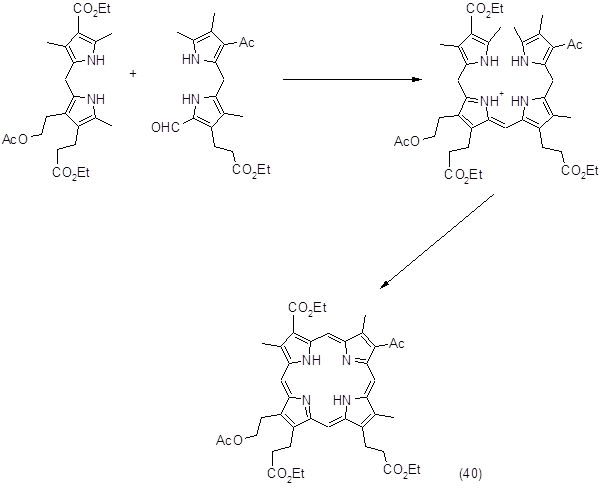
В заключение можно отметить, что использование b‑биленов в качестве промежуточных соединений позволяет получать широкий круг порфиринов с различными заместителями. Среди недостатков этого метода следует отметить недостаточную устойчивость b-биленов и их подверженность нежелательным превращениям.
2.2.2.2. Циклизация а,с-биладиенов.
Биладиены, среди прочих тетрапиррольных структур, играют важную роль в природе (желчные пигменты и протеины) и являются интермедиатами в синтетических подходах к природным порфиринам (65). В 1966 году Джонсон предложил оригинальный метод синтеза порфиринов, который позволяет получать как симметричные, так и несимметричные порфирины (66).
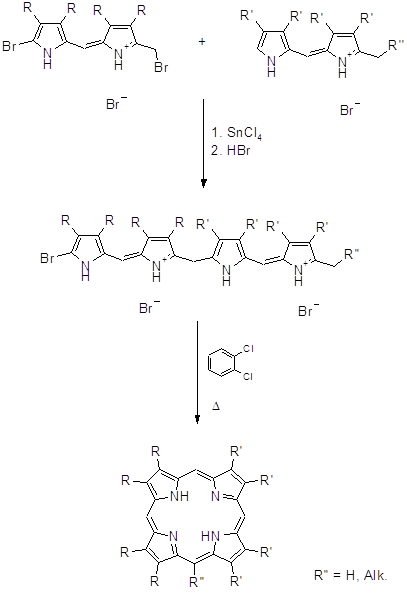
Данный способ нашел применение для алкилзамещенных порфиринов. Выход выше изображенного порфирина на последней стадии составил ~ 70%. На основе а,с- биладиенов также были синтезированы производные мезо-аминопорфирина при аномальной циклизации 1,19-диметил-а,с-диметил-а,с-биладиенов с Cu(OAc)2 в ДМФА, причем мезо-углеродный атом порфиринового макроцикла образуется из карбонильного углерода ДМФА (8,67,68).
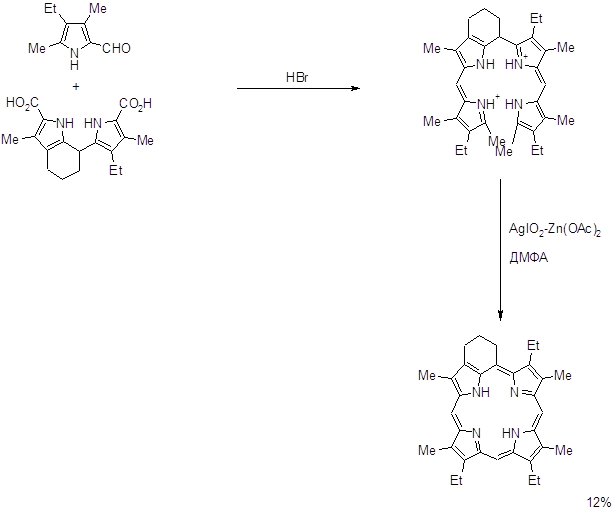
2.2.3. “3+1” порфириновый синтез.
Этот способ позволяет получать как симметричные, так и несимметричные порфирины и их аналоги, причем, выход может достигать отличных для порфиринового синтеза значений ‑ 40-50% (69). Метод состоит в конденсации трипирана с 2,5‑диформилпирролом (70) или 2,5-бис((N,N-диметиламино)метил)пирролом (52,71). Синтез с 2,5‑диформилпирролом используется наиболее часто, его проводят, когда исходные субстраты способны выдержать условия кислотного катализа, либо при однозначном образовании одного изомера порфирина или возможности легкого разделения смеси изомеров. Недостатком выше описанного метода, являлась сложность синтеза 2,5‑диформилпирролов, однако в последние годы был разработан достаточно простой и надежный способ позволяющий получать пиррол дикарбальдегиды с хорошим выходом (6,70,72,73).
Другим возможным вариантом «3+1» порфиринового синтеза является использование 2,5‑бис((N,N‑диметиламино)метил)пиррола, при этом реакция проводится в более мягких условиях, за счет чего достигается образование одного изомера порфирина, а не всех возможных как при использовании 2,5‑диформилпирролов (52). При помощи этого метода стало возможным синтезировать азапорфирины, содержащие в качестве одного из пиррольных циклов имидазольный фрагмент, с удовлетворительным выходом (71).

3. Обсуждение результатов.
Порфириновые макроциклы сопряженные по b-положениям пиррольного кольца с ароматическими и гетероциклическими системами нашли применение в качестве молекулярных зондов, высокоэффективных катализаторов, фотосенсоров в фотодинамической терапии рака и красителей поглощающих в ближнем ИК диапазоне спектра. Целью данной работы является поиск подходов к синтезу сопряженной системы пиррола с гетероциклическим фрагментом для последующего введения ее в порфириновый цикл.
Из литературных источников известно, что существует несколько подходов к синтезу порфиринового макроцикла, наиболее часто используемыми являются методы “2+2” и “3+1. Последний метод был выбран как наиболее подходящий. Для его реализации необходимо было синтезировать b-алкил или b-незамещенный трипиран и пиррол, содержащий в b-положениях функциональные заместители. С этой целью был синтезирован трипиран по следующей схеме:

Алкилирование ацетилацетона проводили по стандартной методике - с применением на первой стадии этилата натрия и последующей обработкой метилиодидом при 400 в течение 1 часа, выход 3–метил–2,4–пентандиона (41) составил 48%. Синтез 2‑карбоэтокси-3,4,5‑триметилпиррола (43) проводили из 3-метил-2,4-пентандиона (41) и изонитрозомалонового эфира (42) конденсацией в уксусной кислоте в присутствии цинка и ацетата натрия при нагревании до 900 в течение 1 часа, выход продукта составил 71%. 2‑ацетоксиметил–3,4–диметил–5–карбоэтоксипиррол (44) получали окислением 2‑карбоэтокси-3,4,5-триметилпиррола (43) тетраацетатом свинца в смеси уксусной кислоты и уксусного ангидрида при комнатной температуре в течение 2 часов, выход 2‑ацетоксиметил–3,4–диметил–5–карбоэтоксипиррола (44) составил 82%. Синтез 1,14-дикарбоэтокси-2,3,12,13-тетраметил трипирана (45) проводили конденсацией в метаноле 1 эквивалента пиррола и 2 эквивалентов 2‑ацетоксиметил–3,4–диметил–5–карбоэтоксипиррола (44) в присутствии толуолсульфокислоты при нагревании до 600 в течение 7 часов, выход 1,14-дикарбоэтокси-2,3,12,13-тетраметил трипирана (45) составил 51%. Все синтезированные продукты были охарактеризованы спектральными методами и были определены их физико-химические константы.
Следующим этапом работы была разработка методов получения монопиррольных интермедиатов для получения сопряженной системы, включающей два гетероциклических фрагмента. Из литературных данных известно, что существует 2 подхода к синтезу сопряженной системы пиррола с гетероциклическим фрагментом:
1. К готовому гетероциклическому фрагменту, используя реакцию Бартона-Зарда, присоединяют пиррольный цикл.
2. К готовому пиррольному циклу, имеющему функциональные группы в b-положениях наращивают гетероциклический фрагмент.
На основе первого подхода было решено провести конденсацию 2-метил-6-нитробензотиазола (47) и этилового эфира изоциануксусной кислоты (51) в присутствии сильных ненуклеофильных оснований, т.е. в условиях реакции Бартона-Зарда (7,20,21).
Нитрование 2-метилбензотиазола (46) проводили нитрующей смесью при нагревании до 900 в течение 5 часов, выход 2-метил-6-нитробензотиазола (47) составил 20%.
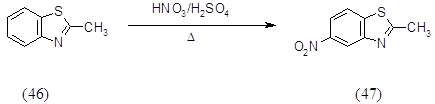
Исходным соединением в синтезе этилового эфира изоциануксусной кислоты (51) являлся глицин (48), который превращали в хлоргидрат глицинэтилового эфира (49) действием тионилхлорида в этаноле при кипячении с обратным холодильником в течение 2 часов. Выход продукта составил 94%. Полученный хлоргидрат глицинэтилового эфира (49) кипятили в этилортоформиате в присутствии толуолсульфокислоты и триэтиламина в течение 20 часов. Получили этиловый эфир N-формилглицина (50) с выходом 66%, который после обработки POCl3 в триэтиламине и дал этиловый эфир изоциануксусной кислоты (51) с выходом 76%. Все синтезированные продукты были охарактеризованы спектральными методами и были определены их физико-химические константы (16).
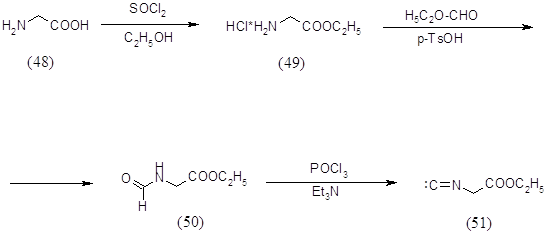
Полученные 2-метил-6-нитробензотиазол (47) и этиловый эфир изоциануксусной кислоты (51) растворяли в абсолютном ТГФ и вводили в конденсацию в присутствии сильных оснований (условия проведения реакций и обработка приведены в таблице). Однако реакция протекала плохо, в основном возвращался исходный 2-метил-6-нитробензотиазол (47) и получалось множество продуктов, суммарный вес которых незначителен.
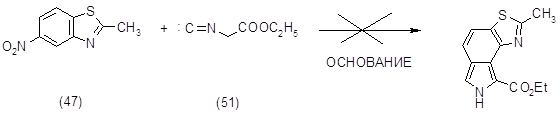
Реакция 2-метил-6-нитробензотиазола и этилового эфира изоциануксусной кислоты.
| Отношение 2‑метил‑6‑нитробензотиазола к этиловому эфиру изоциануксусной кислоты. | Растворитель, объем (на 0,1 г 2-метил-6-нитробензотиазола) (мл) | Основание, отношение к 2‑метил‑6‑нитробензотиазолу | Условия проведения реакции, обработка. |
| 1:1.1 | ТГФ абс. , 50 мл | DBU, 1: 1.1 | 100 часов при комнатной температуре. |
| 1:1.1 | ТГФ абс. , 50 мл | DBU, 1: 1.1 | Кипячение 12 часов. |
| 1:1.1 | ТГФ абс. , 50 мл | (Et2N)3P=NEt, 1:1 | 24 часа при 200 и 4 часа при 600, разбавляют CHCl3 и промывают водой. |
| 1:1.1 | ТГФ абс. , 50 мл | (Et2N)3P=NEt, 1:2 | 200 часов при 200, разбавляют CHCl3 и промывают водой. |
| 1:1.1 | ТГФ абс. , 50 мл | (Et2N)3P=NEt, 1:2 | 200 часов при 500, разбавляют CHCl3 и промывают водой. |
| 1:1.1 | ТГФ абс. , 40 мл | NaH, 1:1 | 72 часа при при 200, разбавляют CHCl3 и промывают водой. |
| 1:1.1 | ТГФ абс. , 40 мл | NaH, 1:2 | 72 часа при при 200, разбавляют CHCl3 и промывают водой. |
| 1:1.1 | ТГФ абс. , 40 мл | NaH, 1:2 | 72 часа при при 500, разбавляют CHCl3 и промывают водой. |
Подобные работы:
© 2010-2021, referat-web.ru