Растворение твердых веществ
Тема контрольной работы «Растворение твердых веществ» по дисциплине «Химическая технология неорганических веществ».
Под термином растворение понимают гетерогенные реакции, протекающие между твердым веществом и жидкостью и сопровождающиеся переходом твердого вещества в раствор. Наиболее часто этот процесс используется в химической промышленности, но он играет важную роль и в металлургической, пищевой промышленности и даже в машино- и приборостроении.
В данной работе рассмотрены основные кинетические закономерности процесса растворения твердых тел, приведена одна из методик экспериментального определения кинетических параметров растворения (константы скорости, энергии активации, порядка реакции) и оценки влияния интенсивности перемешивания на скорость растворения.
I. ОБЩИЕ ПОЛОЖЕНИЯ
1.1 Растворение как гетерогенный химический процесс
Растворение твердых тел – один из важнейших процессов химической технологи. Непосредственный результат растворения заключается в получении раствора, т.е. гомогенной смеси двух и более веществ. Обычно взаимодействие растворителя с полностью растворяющейся твердой фазой происходит на поверхности частиц, в ряде случав это взаимодействие может затрагивать пористую структуру внутри частиц. Можно выделить два основных класса реакций растворения:
1. Обратимое растворение;
2. Необратимое растворение.
Обратимое растворение сводится к образованию сольватов на поверхности реагирующей твердой фазы и переносу их в раствор. Обратимость этого процесса заключается в том, что полученный раствор можно кристаллизацией разделить на исходные реагенты. Примером такого процесса является растворение ионных кристаллов в воде с образованием пересыщенных растворов и их кристаллизация.
Необратимое растворение можно по типу реакций разбить на 3 подгруппы:
а) реакции, сводящиеся к образованию сольватов на поверхности и последующему переносу их в раствор. По своему типу эти реакции могут быть сходными с теми, которые наблюдаются при обратимом растворении. Однако полученный раствор уже нельзя кристаллизацией разделить на исходные компоненты. Примером таких реакций может служить растворение смешанных кристаллов, состоящих из ионных молекул, либо стекол в полярных и неполярных жидкостях;
б) окислительно-восстановительные реакции, приводящие к образованию сольватированных ионов и продуктов восстановления окислителя. К такого рода реакциям относятся взаимодействие металлов или их сплавов с окислителями или комплексообразователями в водных растворах;
в) реакции присоединения, замещения, нейтрализации. К этой подгруппе относятся реакции взаимодействия молекулярных и ионных кристаллов с полярными и неполярными жидкостями, приводящие к образованию сольватированных молекул и ионов.
Все эти реакции объединяет общее для гетерогенных процессов свойство: реакции растворения всегда включают в себя несколько стадий. Первой из них является стадия переноса растворителя к поверхности растворяющегося вещества, на которой происходит реакция; на второй стадии происходит собственно химическая реакция; третья стадия заключается в отводе продуктов реакции от реакционной поверхности.
Суммарная скорость процесса растворения определяется скоростями отдельных стадий. Если скорость одной из стадий процесса существенно меньше, чем скорость других, суммарная скорость процесса будет определяться скоростью именно этой наиболее медленной стадии. В том случае, когда медленной (лимитирующей) стадией является химическое взаимодействие, концентрация реагирующего вещества у поверхности совпадает с концентрацией в объеме, а наблюдаемая скорость реакции зависит от внешних параметров (т.е. от температуры и концентрации) точно также, как и истинная скорость реакции на поверхности. Такую предельную область гетерогенного процесса принято называть кинетической.
Если же медленной стадией процесса является подвод реагентов к поверхности или отвод продуктов реакции от поверхности, то скорость процесса определяется скоростью диффузии, и макроскопическая кинетика реакции не имеет ничего общего с истинной кинетикой на поверхности. Эту предельную область гетерогенного процесса называют диффузионной.
Наконец, если скорости отдельных стадий сравнимы между собой, то процесс протекает в переходной области, и наблюдаемая скорость процесса определяется как скоростью диффузии, так и скоростью химической реакции на поверхности растворяющегося вещества.
1.2 Уравнения кинетики растворения
Общие уравнения диффузионно-кинетического режима получены Д.А. Франк-Каменецким в предположении, что условия диффузионного транспорта вещества могут приближенно считаться не зависящими от условий протекания химической реакции на поверхности. Это предположение справедливо в том случае, когда все участки поверхности можно считать одинаково доступными в диффузионном отношении (так называемая равнодоступная поверхность). Скорость реакции на поверхности ![]() пропорциональна концентрации активного реагента С у поверхности в некоторой степени
пропорциональна концентрации активного реагента С у поверхности в некоторой степени ![]() , определяющей формальный порядок реакции:
, определяющей формальный порядок реакции:
![]() (1)
(1)
Диффузионный поток к поверхности можно определить с помощью коэффициента массоотдачи ![]() :
:
![]() (2)
(2)
где ![]() - концентрация реагента в объеме.
- концентрация реагента в объеме.
В стационарном состоянии количество вещества, вступающее в реакцию на поверхности, равно диффузионному потоку:
![]() (3)
(3)
Уравнение (3) является общим уравнением диффузионно-кинетического режима. При ![]() , т.е. когда реакция имеет первый порядок, получаем:
, т.е. когда реакция имеет первый порядок, получаем:
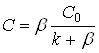 (4)
(4)
Для квазистационарной скорости реакции получим при ![]() :
:
![]() (5)
(5)
где 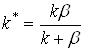 . (6)
. (6)
Это соотношение принимает особенно наглядный вид, если вместо константы скорости реакции и коэффициента массоотдачи рассматривать обратные им величины:
 (7)
(7)
Таким образом, полное сопротивление диффузионно-кинетического процесса равно сумме кинетического и диффузионного сопротивлений.
В случае обратимой реакции у поверхности устанавливается концентрация, соответствующая термодинамическому равновесию. Поэтому для простых реакций растворения, сводящихся лишь к образованию на поверхности сольватов и отводу их в раствор, предельный диффузионный поток определяется выражением:
![]()
где ![]() - концентрация насыщенного раствора,
- концентрация насыщенного раствора,
![]() - концентрация растворяемого компонента в объеме.
- концентрация растворяемого компонента в объеме.
Следовательно, в диффузионной области роль константы скорости реакции играет коэффициент массоотдачи ![]() , и наблюдаемая скорость процесса не имеет ничего общего с истинной кинетикой химической реакции.
, и наблюдаемая скорость процесса не имеет ничего общего с истинной кинетикой химической реакции.
Коэффициент массоотдачи ![]() зависит не только от физических свойств раствора, но и от гидродинамических условий взаимодействия частицы с окружающей её сплошной средой. К сожалению, даже в тех случаях, когда межфазовая поверхность может приблизительно рассматриваться как равнодоступная, количественная оценка с помощью уравнений (3) и (4) не может быть дана. Во - первых, мы не располагаем надежными данными для того, чтобы судить об интенсивности конвективной диффузии к частицам, взвешенным в растворе с помощью перемешивающего устройства. Во – вторых, межфазная поверхность совокупности растворяющихся полидисперсных частиц произвольной формы может быть оценена весьма приблизительно, особенно при степени растворения выше 10-20%. Кроме того, в процессе растворения удельная внешняя поверхность также является переменной величиной: функционально уменьшается во времени в зависимости от геометрического размера (диаметра) растворяющихся частиц.
зависит не только от физических свойств раствора, но и от гидродинамических условий взаимодействия частицы с окружающей её сплошной средой. К сожалению, даже в тех случаях, когда межфазовая поверхность может приблизительно рассматриваться как равнодоступная, количественная оценка с помощью уравнений (3) и (4) не может быть дана. Во - первых, мы не располагаем надежными данными для того, чтобы судить об интенсивности конвективной диффузии к частицам, взвешенным в растворе с помощью перемешивающего устройства. Во – вторых, межфазная поверхность совокупности растворяющихся полидисперсных частиц произвольной формы может быть оценена весьма приблизительно, особенно при степени растворения выше 10-20%. Кроме того, в процессе растворения удельная внешняя поверхность также является переменной величиной: функционально уменьшается во времени в зависимости от геометрического размера (диаметра) растворяющихся частиц.
К реакциям, происходящим в пористом материале выводы, полученные для равнодоступной поверхности, совершенно неприменимы.
Рассмотрение диффузионной кинетики дано в работах Я.Б. Зельдовича.
Для стационарного процесса макроскопическая скорость реакции определяется уравнением:
 (8)
(8)
С точностью до безразмерного множителя порядка единицы глубину проникновения реакции L можно определить так:
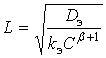 (9)
(9)
Для квазистационарного процесса диффузионный поток равен наблюдаемой скорости реакции. Поэтому:
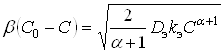 , (10)
, (10)
где ![]() - диффузионный поток активного реагента из объема жидкой фазы к поверхности твердого вещества.
- диффузионный поток активного реагента из объема жидкой фазы к поверхности твердого вещества.
Решая уравнение (10) относительно С и подставляя полученное значение в (8) при ![]() , получим:
, получим:
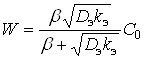 (11)
(11)
При растворении пористого материала или в случае, когда один из компонентов твердого тела не растворяется, а второй - растворяется возможны четыре предельные области:
1. При ![]() скорость суммарного процесса определяется диффузией в объеме и совпадает с предельным диффузионным потоком к равнодоступной поверхности, равной внешней поверхности пористого материала:
скорость суммарного процесса определяется диффузией в объеме и совпадает с предельным диффузионным потоком к равнодоступной поверхности, равной внешней поверхности пористого материала: ![]() . Концентрация активного реагента даже на поверхности пористого материала, а тем более внутри пор растворяющегося вещества, гораздо меньше, чем в объеме. Эту предельную область называют внешней диффузионной.
. Концентрация активного реагента даже на поверхности пористого материала, а тем более внутри пор растворяющегося вещества, гораздо меньше, чем в объеме. Эту предельную область называют внешней диффузионной.
2. При ![]() и
и ![]() (Н – полная толщина пористого слоя; L – глубина проникновения реакции, вычисленная по формуле (9); rn - средний диаметр пор) определяющей стадией является диффузия в порах. Концентрация активного реагента на поверхности растворяющегося вещества близка к концентрации в объеме жидкой фазы, но по мере удаления от поверхности пористого материала в глубину пор она снижается практически до нуля. Эта область называется внутридиффузионной.
(Н – полная толщина пористого слоя; L – глубина проникновения реакции, вычисленная по формуле (9); rn - средний диаметр пор) определяющей стадией является диффузия в порах. Концентрация активного реагента на поверхности растворяющегося вещества близка к концентрации в объеме жидкой фазы, но по мере удаления от поверхности пористого материала в глубину пор она снижается практически до нуля. Эта область называется внутридиффузионной.
3. ![]() и
и ![]() концентрация активного реагента в порах твердого вещества совпадает с концентрацией в объеме жидкой фазы. Такая ситуация возникает, если пористый материал черезвычайно доступен в диффузионном отношении (вещества с большим диаметром пор, а коэффициент диффузии в объеме жидкой фазы имеет тот же порядок, что и коэффициент диффузии в порах) и в то же время обладает относительно низкой химической активностью по отношению к растворителю.
концентрация активного реагента в порах твердого вещества совпадает с концентрацией в объеме жидкой фазы. Такая ситуация возникает, если пористый материал черезвычайно доступен в диффузионном отношении (вещества с большим диаметром пор, а коэффициент диффузии в объеме жидкой фазы имеет тот же порядок, что и коэффициент диффузии в порах) и в то же время обладает относительно низкой химической активностью по отношению к растворителю.
В этой внутрикинетической области вся поверхность пористого материала взаимодействует с активным реагентом при одной и той же концентрации ![]() и микроскопическая скорость реакции пропорциональна объему пористого материала.
и микроскопическая скорость реакции пропорциональна объему пористого материала.
4. При ![]() и
и ![]() реакция протекает только на внешней поверхности пористого материала. Это означает, что вследствие относительно высокой химической активности или малой пористости (вещества с очень малым диаметром пор, т.е когда диаметр пор и диаметр молекул растворителя или растворенного вещества имеют один порядок) реагент не проникает в глубь пористого материала. В то же время предельный диффузионный поток из объема к внешней поверхности твердого вещества во много раз больше скорости химической реакции. Такая область называется внешнекинетической. Микроскопическая скорость в этой области пропорциональна внешней поверхности пористого материала.
реакция протекает только на внешней поверхности пористого материала. Это означает, что вследствие относительно высокой химической активности или малой пористости (вещества с очень малым диаметром пор, т.е когда диаметр пор и диаметр молекул растворителя или растворенного вещества имеют один порядок) реагент не проникает в глубь пористого материала. В то же время предельный диффузионный поток из объема к внешней поверхности твердого вещества во много раз больше скорости химической реакции. Такая область называется внешнекинетической. Микроскопическая скорость в этой области пропорциональна внешней поверхности пористого материала.
II. ЭКСПЕРИМЕНТАЛЬНОЕ ОПРЕДЕЛЕНИЕ КИНЕТИЧЕСКИХ ХАРАКТЕРИСТИК
2.1 Определение энергии активации
При проведении кинетических исследований следует убедиться в безградиентности процесса: повышение интенсивности перемешивания не приводит к росту константы скорости реакции.
Для определения энергии активации необходимо провести не менее двух периодических опытов при различных температурах. В каждом из опытов при фиксированных температурах ![]() и
и ![]() (
(![]() должно быть не менее 100 С для уменьшения погрешности в численном значении энергии активации Е) определяют зависимость доли нерастворившегося компонента от продолжительности растворения:
должно быть не менее 100 С для уменьшения погрешности в численном значении энергии активации Е) определяют зависимость доли нерастворившегося компонента от продолжительности растворения: ![]() и
и ![]() ,
, ![]() (
(![]() - степень растворения вещества). При этом время растворения вещества
- степень растворения вещества). При этом время растворения вещества ![]() и концентрация растворителя во всех опытах должно быть одинаковым или необходимо фиксировать время достижения одинаковых степеней растворения вещества, а все остальные параметры системы должны остаться неизменными.
и концентрация растворителя во всех опытах должно быть одинаковым или необходимо фиксировать время достижения одинаковых степеней растворения вещества, а все остальные параметры системы должны остаться неизменными.
Кинетические функции растворения могут быть представлены в виде:
 (12)
(12)
Если сравнить скорость растворения при равных значениях ![]() , то
, то ![]() и
и ![]() (т.к. начальные концентрации одинаковы исходя из начально заданных условий, а изменение концентрации в ходе растворения стехиометрически связано с долей растворившегося продукта). Поэтому для равных значений
(т.к. начальные концентрации одинаковы исходя из начально заданных условий, а изменение концентрации в ходе растворения стехиометрически связано с долей растворившегося продукта). Поэтому для равных значений ![]() можно записать:
можно записать:
 (13)
(13)
Отношение, стоящее в правой части уравнения (13), не зависит от времени.
Интегрируя это уравнение и подставляя вместо ![]() и
и ![]() их значения, равные
их значения, равные ![]() и
и ![]() , получим окончательно:
, получим окончательно:
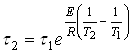 (14)
(14)
Необходимо подчеркнуть, что ![]() и
и ![]() означают время, необходимое для достижения одного и того же значения
означают время, необходимое для достижения одного и того же значения ![]() в первом и втором опытах.
в первом и втором опытах.
Уравнение (14) показывает, что зависимость ![]() от
от ![]() выражается аппроксимирующей линией, тангенс угла наклона которой к оси абсцисс численно равен энергии активации Е/R.
выражается аппроксимирующей линией, тангенс угла наклона которой к оси абсцисс численно равен энергии активации Е/R.
Вместо определения углового коэффициента прямой, энергию активации можно вычислить по формуле:
 (15)
(15)
Вычисления по формуле (15) в других интервалах ![]() дадут набор значений Е, из которого легко получить наиболее достоверное (среднее) значение энергии активации и среднюю ошибку её определения, которая не должна быть более 10%. В противном случае необходимо искать ошибку в постановке эксперимента или в расчетах. Возможно также изменение механизма процесса растворения. В последнем случае необходимо сузить температурные интервалы и увеличить количество экспериментов.
дадут набор значений Е, из которого легко получить наиболее достоверное (среднее) значение энергии активации и среднюю ошибку её определения, которая не должна быть более 10%. В противном случае необходимо искать ошибку в постановке эксперимента или в расчетах. Возможно также изменение механизма процесса растворения. В последнем случае необходимо сузить температурные интервалы и увеличить количество экспериментов.
2.2 Определение порядка реакции
химический процесс растворение
Для определения порядка реакции нужно провести не менее двух периодических опытов при одной и той же температуре, но при различных начальных концентрациях активного реагента ![]() ≠
≠![]() . В этих опытах определяют зависимости доли нерастворившегося компонента от продолжительности растворения
. В этих опытах определяют зависимости доли нерастворившегося компонента от продолжительности растворения ![]() и
и ![]() . Кроме того, необходима информация об изменении концентрации активного реагента в ходе растворения, т.е. зависимости
. Кроме того, необходима информация об изменении концентрации активного реагента в ходе растворения, т.е. зависимости ![]() и
и ![]() , где
, где ![]() и
и ![]() - текущие концентрации активного реагента в первом и втором опытах. Эти зависимости могут быть получены либо прямыми измерениями (в тех же опытах), либо расчетным путем, исходя из стехиометрических соотношений. При этом необходимо учитывать, что данные полученные расчетным путем дают, как правило большую ошибку, чем экспериментальные данные (при условии грамотной постановки эксперимента). Затем по экспериментальным данным строят зависимость
- текущие концентрации активного реагента в первом и втором опытах. Эти зависимости могут быть получены либо прямыми измерениями (в тех же опытах), либо расчетным путем, исходя из стехиометрических соотношений. При этом необходимо учитывать, что данные полученные расчетным путем дают, как правило большую ошибку, чем экспериментальные данные (при условии грамотной постановки эксперимента). Затем по экспериментальным данным строят зависимость ![]() от
от ![]() , где
, где ![]() и
и ![]() отвечают равным значениям
отвечают равным значениям ![]() и
и ![]() . Из уравнений для скорости растворения (12) при равных значениях
. Из уравнений для скорости растворения (12) при равных значениях ![]() следует соотношение:
следует соотношение:
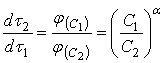 (16)
(16)
откуда
 (16а)
(16а)
Уравнение (16) относится к произвольному, но фиксированному значению ![]() . Отношение приращений
. Отношение приращений ![]() и текущие концентрации активного реагента
и текущие концентрации активного реагента ![]() и
и ![]() должны быть определены в точках, отвечающих этим значениям. Как и в случае определения энергии активации по уравнениям (14) и (15) вычисления по уравнению (16) или (16а) можно выполнить для целого ряда значений
должны быть определены в точках, отвечающих этим значениям. Как и в случае определения энергии активации по уравнениям (14) и (15) вычисления по уравнению (16) или (16а) можно выполнить для целого ряда значений ![]() и за тем усреднить полученные результаты. При этом необходимо помнить, что расхождения результатов параллельных изменений не должны превышать 10%.
и за тем усреднить полученные результаты. При этом необходимо помнить, что расхождения результатов параллельных изменений не должны превышать 10%.
Из изложенного выше следует, что определение порядка реакции много сложнее чем определение энергии активации, так как требует численного или графического дифференцирования полученной из опытов зависимости ![]() . Причина этого усложнения ясна: концентрация активного реагента в периодическом опыте, как правило, может изменяться в больших интервалах. В тоже время температуру в ходе опыта можно легко поддерживать постоянной, в пределах
. Причина этого усложнения ясна: концентрация активного реагента в периодическом опыте, как правило, может изменяться в больших интервалах. В тоже время температуру в ходе опыта можно легко поддерживать постоянной, в пределах ![]() . Определение порядка реакции можно упростить, если концентрацию активного реагента в ходе периодического опыта не изменять. Это достигается путем восполнения убыли реагента в ходе растворения (периодическое дозирование), или сделать это изменение пренебрежимо малым, проводя опыт с очень большим избытком активного реагента. Этого можно достичь предварительным расчетом, чтобы даже при полном растворении твердого растворяющегося вещества, концентрация растворителя не снизилась более, чем на 5-10% от исходной. Во всех выше описанных случаях правая часть уравнения (16) или (16а) должна являться постоянной величиной, поэтому
. Определение порядка реакции можно упростить, если концентрацию активного реагента в ходе периодического опыта не изменять. Это достигается путем восполнения убыли реагента в ходе растворения (периодическое дозирование), или сделать это изменение пренебрежимо малым, проводя опыт с очень большим избытком активного реагента. Этого можно достичь предварительным расчетом, чтобы даже при полном растворении твердого растворяющегося вещества, концентрация растворителя не снизилась более, чем на 5-10% от исходной. Во всех выше описанных случаях правая часть уравнения (16) или (16а) должна являться постоянной величиной, поэтому 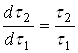 , и зависимость
, и зависимость ![]() является линейной.
является линейной.
Поэтому необходимость в дифференцировании отпадает, и расчетная формула для ![]() принимает вид:
принимает вид:
 (17)
(17)
причем ![]() и
и ![]() - теперь уже постоянные величины.
- теперь уже постоянные величины.
2.3 Определение кинетической функции
Для описания растворения необходима кинетическая характеристика процесса растворения. Выразим время в безразмерных единицах – в долях времени полного растворения ![]() . Безразмерное время, равное отношению продолжительности растворения
. Безразмерное время, равное отношению продолжительности растворения ![]() к времени полного растворения
к времени полного растворения ![]() , обозначим через х:
, обозначим через х:
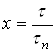 .
.
В качестве кинетической характеристики процесса удобно использовать зависимость доли нерастворившегося компонента ![]() от безразмерного времени х при постоянных концентрациях и температуре. Зависимость
от безразмерного времени х при постоянных концентрациях и температуре. Зависимость ![]() называется кинетической функцией.
называется кинетической функцией.
Результаты периодического опыта дают зависимость доли нерастворившегося продукта ![]() от времени
от времени ![]() , для получения кинетической функции
, для получения кинетической функции ![]() достаточно выразить время в безразмерных единицах , причем время полного растворения
достаточно выразить время в безразмерных единицах , причем время полного растворения ![]() определяют в том же опыте.
определяют в том же опыте.
Подобный способ с известным приближением можно применять в тех случаях, когда изменения концентрации активного реагента в ходе растворения малы (например при большом избытке активного реагента).
Для определения кинетической функции могут быть использованы результаты любого периодического опыта, проведенного при постоянных значениях концентрации активного реагента и температуры. Методика определения сводиться к следующему.
1. Результаты периодического опыта представляют в виде зависимостей доли нерастворившегося компонента ![]() и концентрации активного реагента
и концентрации активного реагента ![]() от времени
от времени ![]() :
: ![]() и
и ![]() .
.
Эти зависимости можно представить в виде графиков, таблиц или уравнений, в соответствии с выбранным способом вычисления интегралов. Безразмерное время х, соответствующее любому значению ![]() рассчитывают, пользуясь уравнением:
рассчитывают, пользуясь уравнением:
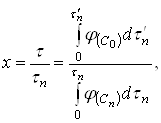 (18)
(18)
где ![]() - время, необходимое для достижения любого значения
- время, необходимое для достижения любого значения ![]() в условиях периодического опыта, т.е. при переменной концентрации активного реагента
в условиях периодического опыта, т.е. при переменной концентрации активного реагента ![]() .
.
Верхние пределы интегрирования 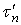 и
и 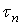 выражают соответственно время, необходимое для достижения определенного значения
выражают соответственно время, необходимое для достижения определенного значения 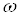 и время полного растворения в периодическом опыте с изменяющейся концентрацией реагента (в отличие от
и время полного растворения в периодическом опыте с изменяющейся концентрацией реагента (в отличие от 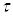 и , выражающих те же понятия, но при постоянной концентрации С.
и , выражающих те же понятия, но при постоянной концентрации С.
2. По уравнению (18) вычисляют значение х, отвечающее определенному значению ![]() .
.
При этом верхний предел интеграла, стоящий в числителе, означает время необходимое для достижения этого значения ![]() в периодическом опыте. Такие вычисления проводят для ряда последовательных значений доли нерастворившегося компонента
в периодическом опыте. Такие вычисления проводят для ряда последовательных значений доли нерастворившегося компонента ![]() . Результаты расчетов, представленные в виде графика, таблицы или апроксимированные аналитическим выражением, дают кинетическую функцию
. Результаты расчетов, представленные в виде графика, таблицы или апроксимированные аналитическим выражением, дают кинетическую функцию ![]()
Графический вариант описанного метода иллюстрируется рис.1
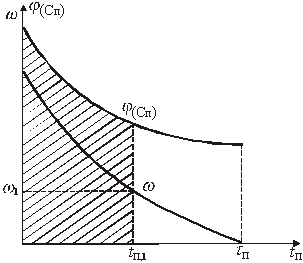
Рис.1. К графическому определению кинетической функции
Значение х соответствующее некоторому ![]() , равно отношению заштрихованной площади ко всей площади под кривой
, равно отношению заштрихованной площади ко всей площади под кривой ![]() . С помощью такого графика можно легко определить значение х для ряда значений
. С помощью такого графика можно легко определить значение х для ряда значений ![]() и составить таблицу или построить график зависимости
и составить таблицу или построить график зависимости ![]() .
.
Из уравнения (18) видно, что для определения х нужно знать зависимость скорости процесса от концентрации, т.е. функцию 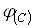 .
.
Если, например  , то должен быть известен порядок реакции
, то должен быть известен порядок реакции 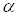 . Тогда уравнение (18) принимает вид:
. Тогда уравнение (18) принимает вид:
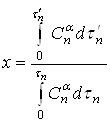 (19)
(19)
Входящие в уравнение (18) и (19) интегралы могут быть определены численными или графическими методами. Необходимая для выполнения интегрирования зависимость ![]() от
от ![]() , а также связь между
, а также связь между ![]() и
и ![]() , содержатся в результатах периодического опыта.
, содержатся в результатах периодического опыта.
Таким образом, уравнение (18) позволяет вычислить значения, отвечающие любому значению ![]() , и тем самым определить кинетическую функцию.
, и тем самым определить кинетическую функцию.
2.4 Определение времени полного растворения
В отличие от кинетической функции 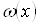 , которая сохраняет свой вид для любых постоянных значений Т и С, величина всегда относится к совершенно определенным условиям растворения и изменяется при изменении этих условий. Очевидно, что если растворение проходит при постоянной концентрации активного реагента, то величина совпадает со временем полного растворения в периодическом опыте:
, которая сохраняет свой вид для любых постоянных значений Т и С, величина всегда относится к совершенно определенным условиям растворения и изменяется при изменении этих условий. Очевидно, что если растворение проходит при постоянной концентрации активного реагента, то величина совпадает со временем полного растворения в периодическом опыте: 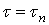 . При переменной концентрации дело обстоит иначе: величина связана с отношением:
. При переменной концентрации дело обстоит иначе: величина связана с отношением:
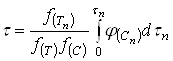 (20)
(20)
В частности, для определения времени полного растворения при некоторых фиксированных значениях ![]() и
и ![]() достаточно подставить в уравнение (20)
достаточно подставить в уравнение (20) ![]() вместо
вместо ![]() и
и ![]() вместо
вместо ![]() :
:
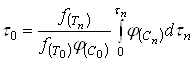 (21)
(21)
Естественно использовать для определения ![]() тот периодический опыт, для которого
тот периодический опыт, для которого ![]() . Тогда формула (21) упрощается:
. Тогда формула (21) упрощается:
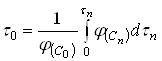 (22)
(22)
Входящий в уравнения (21) и (22) интеграл численно равен площади под кривой ![]() на рис.1. Для его вычисления можно воспользоваться любым из известных методов.
на рис.1. Для его вычисления можно воспользоваться любым из известных методов.
Таким образом, результаты периодического опыта, проведенного при изменяющейся концентрации активного реагента ![]() , позволяют легко определить время полного растворения
, позволяют легко определить время полного растворения ![]() , относящейся к постоянным значениям
, относящейся к постоянным значениям ![]() и
и ![]() . Для перехода к иным значениям концентрации и температуры можно воспользоваться соотношением, вытекающим из уравнений (20) и (21).
. Для перехода к иным значениям концентрации и температуры можно воспользоваться соотношением, вытекающим из уравнений (20) и (21).
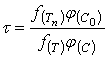 (23)
(23)
В частности, если ![]() и
и ![]() , то:
, то:
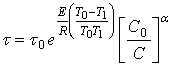 (24)
(24)
Таким образом, зная энергию активации Е, порядок реакции ![]() и время полного растворения
и время полного растворения ![]() при некоторых фиксированных значениях
при некоторых фиксированных значениях ![]() и
и ![]() по формуле (24) можно вычислить время полного растворения
по формуле (24) можно вычислить время полного растворения ![]() при любых значениях
при любых значениях ![]() и
и ![]() .
.
2.5 Рекомендации по проведению исследований и выполнению расчетов
Результаты одного лабораторного периодического опыта в принципе позволяют определить кинетическую функцию ![]() . В действительности нужно провести серию опытов в диапазоне интересующих значений концентрации активного реагента и температуры. Это необходимо для обеспечения надежности кинетических величин.
. В действительности нужно провести серию опытов в диапазоне интересующих значений концентрации активного реагента и температуры. Это необходимо для обеспечения надежности кинетических величин.
Кроме того, проведение серии опытов необходимо для экспериментального подтверждения инвариантности кинетической функции относительно концентрации и температуры.
Результатом такой серии опытов будет совокупность кривых ![]() . Из каждой кривой нужно определить кинетическую функцию, вычислив для ряда значений
. Из каждой кривой нужно определить кинетическую функцию, вычислив для ряда значений ![]() соответствующие значения х по формуле (18). Из этой формулы следует, что для расчета нужно знать время полного растворения
соответствующие значения х по формуле (18). Из этой формулы следует, что для расчета нужно знать время полного растворения ![]() в периодическом опыте и иметь достаточно надежные данные о ходе кинетической кривой во всем диапазоне значений
в периодическом опыте и иметь достаточно надежные данные о ходе кинетической кривой во всем диапазоне значений ![]() от 0 до
от 0 до ![]() .
.
Аргумент кинетической функции х есть отношение продолжительности растворения и времени полного растворения . Величина играет, следовательно, роль нормировочного множителя или масштабного коэффициента, позволяющего выразить время в безразмерных единицах. В качестве такого коэффициента с равным успехом может быть использовано время, необходимое для достижения любого фиксированного значения . Пусть это фиксированное значение равно 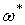 , оно должно быть выбрано таким образом, чтобы изменению от 1 до соответствовали достаточно надежные участки всех экспериментальных кривых. Время, необходимое для достижения значения при постоянных температуре и концентрации активного реагента, обозначим через
, оно должно быть выбрано таким образом, чтобы изменению от 1 до соответствовали достаточно надежные участки всех экспериментальных кривых. Время, необходимое для достижения значения при постоянных температуре и концентрации активного реагента, обозначим через 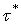 . Введем теперь новое безразмерное время
. Введем теперь новое безразмерное время  :
:  . При обработке экспериментальных данных относящихся к каждому проведенному опыту, вместо определения х по формуле (18) вычисляют значения по формуле:
. При обработке экспериментальных данных относящихся к каждому проведенному опыту, вместо определения х по формуле (18) вычисляют значения по формуле:
 (25)
(25)
где ![]() -время, необходимое для достижения
-время, необходимое для достижения ![]() в периодическом опыте.
в периодическом опыте.
В результате такой обработки экспериментальных данных получают зависимость доли нерастворившегося компонента ![]() от безразмерного времени
от безразмерного времени
![]() при постоянных концентрации активного реагента и температуре. Нетрудно убедиться в том, что, зависимость
при постоянных концентрации активного реагента и температуре. Нетрудно убедиться в том, что, зависимость ![]() есть полный аналог кинетической функции
есть полный аналог кинетической функции ![]() . Аргументы х и
. Аргументы х и ![]() отличаются друг от друга лишь постоянным множителем.
отличаются друг от друга лишь постоянным множителем.
Действительно, из соотношений 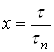 и
и  следует, что
следует, что
 (26)
(26)
где 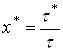 - значение безразмерного времени х, отвечающее выбранной фиксированной величине
- значение безразмерного времени х, отвечающее выбранной фиксированной величине ![]() . Соотношение (26) показывает, что зависимость
. Соотношение (26) показывает, что зависимость ![]() сохраняет присущее кинетической функции свойство инвариантности относительно условий проведения процесса (поскольку величины х и
сохраняет присущее кинетической функции свойство инвариантности относительно условий проведения процесса (поскольку величины х и ![]() не зависят от этих условий).
не зависят от этих условий).
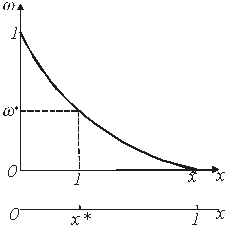
Переход от функции 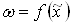 к кинетической функции
к кинетической функции 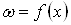 эквивалентен простому изменению масштаба по оси абцисс. Это поясняется рисунком 2, где обе зависимости
эквивалентен простому изменению масштаба по оси абцисс. Это поясняется рисунком 2, где обе зависимости 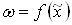 и - изображаються одной и той же кривой и различаются только масштабом по оси абцисс. В отличие от х, изменяющегося от 0 до 1, аргумент зависимости изменяется от 0 до некоторого значения
и - изображаються одной и той же кривой и различаются только масштабом по оси абцисс. В отличие от х, изменяющегося от 0 до 1, аргумент зависимости изменяется от 0 до некоторого значения 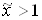 .
.
![]()
![]() Рис.2. Зависимость доли нерастворившегося компонента
Рис.2. Зависимость доли нерастворившегося компонента ![]() от безразмерного времени
от безразмерного времени ![]() .
.
На рис.2 безразмерное время ![]() пропорционально отношению
пропорционально отношению ![]() , где
, где ![]() - время, необходимое для достижения некоторого фиксированного значения
- время, необходимое для достижения некоторого фиксированного значения ![]() .
.
Для получения кинетической функции достаточно разделить все значения на этот постоянный коэффициент  . Предлагаемое видоизменение методики определения кинетической функции сводится к следующему:
. Предлагаемое видоизменение методики определения кинетической функции сводится к следующему:
1. Результаты каждого периодического опыта необходимо представить в виде зависимости доли нерастворившегося компонента ![]() и концентрации активного реагента
и концентрации активного реагента ![]() от времени
от времени ![]() :
: ![]() и
и ![]() .
.
2. Выбирают некоторое фиксированное значение ![]() с таким расчетом, чтобы значению
с таким расчетом, чтобы значению ![]() от 1 до
от 1 до ![]() соответствовали достаточно надежные участки всех экспериментальных кривых.
соответствовали достаточно надежные участки всех экспериментальных кривых.
3. По уравнению (25) вычисляют значение ![]() , отвечающие ряду последовательных значений
, отвечающие ряду последовательных значений ![]() . Такие расчеты выполняют для каждого опыта.
. Такие расчеты выполняют для каждого опыта.
4. Результаты расчетов используют для получения усредненной по всем опытам зависимости ![]() . Эту зависимость удобно нанести на график вместе с результатами обработки отдельных опытов и тем самым убедиться в инвариантности функции
. Эту зависимость удобно нанести на график вместе с результатами обработки отдельных опытов и тем самым убедиться в инвариантности функции ![]() относительно технологических параметров процесса в исследованном диапазоне.
относительно технологических параметров процесса в исследованном диапазоне.
5. С помощью формулы:
 (27)
(27)
необходимо перейти к обычной кинетической функции ![]() т.е. необходимо изменить масштаб по оси абцисс (рис.2).
т.е. необходимо изменить масштаб по оси абцисс (рис.2).
Последний этап необходим для единообразия вычислительных процессов при переходе к их моделированию. Возможные значения безразмерного времени должны быть заключены в диапазон от 0 до 1. Поэтому использование кинетической функции ![]() , в которой аргумент нормирования в максимальной продолжительности процесса - времени полного растворения, предпочтительнее.
, в которой аргумент нормирования в максимальной продолжительности процесса - времени полного растворения, предпочтительнее.
Заметим, что при растворении труднорастворимых веществ и выщелачивании чаще всего не достигают стопроцентного растворения или извлечения полезного компонента. В этом случае под временем полного растворения ![]() естественно понимать время достижения максимального возможного извлечения. Если обозначить максимально возможное извлечение через
естественно понимать время достижения максимального возможного извлечения. Если обозначить максимально возможное извлечение через ![]() , то связь между долей нерастворившегося компонента и достигнутым извлечением
, то связь между долей нерастворившегося компонента и достигнутым извлечением ![]() опишется формулой:
опишется формулой:
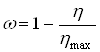 (28)
(28)
Это значит, что при определении ![]() не следует принимать во внимание ту часть полезного компонента, которая не может быть переведена в раствор. Тогда изменению
не следует принимать во внимание ту часть полезного компонента, которая не может быть переведена в раствор. Тогда изменению ![]()