Хроматографический анализ
Хроматография - это физико-химический метод разделения и анализа смесей газов, паров, жидкостей или растворенных веществ сорбционными методами в динамических условиях. Метод основан на различном распределении веществ между двумя несмешивающимися фазами - подвижной и неподвижной.
Подвижной фазой может быть жидкость или газ, неподвижной фазой - твердое вещество, которое называют носителем. При движении подвижной фазы вдоль неподвижной, компоненты смеси сорбируются на неподвижной фазе. Каждый компонент сорбируется в соответствии со сродством к материалу неподвижной фазы (вследствие адсорбции или других механизмов). Поэтому неподвижную фазу называют такжесорбентом. Захваченные сорбентом молекулы могут перейти в подвижную фазу и продвигаться с ней дальше, затем снова сорбироваться.
Таким, образом,хроматографию можно определить как процесс, основанный на многократном повторении актов сорбции и десорбции вещества при перемещении его в потоке подвижной фазы вдоль неподвижного сорбента. Чем сильнее сродство компонента к неподвижной фазе, тем сильнее он сорбируется и дольше задерживается на сорбенте; тем медленнее его продвижение вместе с подвижной фазой. Поскольку компоненты смеси обладают разным сродством к сорбенту, при перемещении смеси вдоль сорбента произойдет разделение: одни компоненты задержатся в начале пути, другие продвинутся дальше. В хроматографическом процессе сочетаются термодинамический (установление равновесия между фазами) и кинетический (движение компонентов с разной скоростью) аспекты.
1. История вопроса
Хроматографический метод анализа разработан русским ботаником М.С.Цветом в 1903 г. С помощью этого метода ему удалось разделить хлорофилл на составляющие окрашенные вещества. При пропускании экстракта хлорофилла через колонку, заполненную порошком мела, и промывании петролейным эфиром он получил несколько окрашенных зон и назвал эти зоны хроматограммой (от греческого “хроматос” — цвет), а метод - хроматографией. Н.А.Измайлов и М.С.Шрайбер в 1938 г. разработали новый вид хроматографии, получивший название тонкослойной. Ими были разделены алкалоиды, экстрагированные из лекарственных растений на оксиде алюминия, нанесенном на стекло.
Отправной точкой бурного развития многих методов хроматографического анализа является работа лауреатов Нобелевской премии A.Мартина и Р.Синджа, ими был предложен и разработан метод распределительной хроматографии (1941г.). В 1952 г. А.Мартином и Л.Джеймсом были получены первые результаты в области газожидкостной хроматографии. Эти работы вызвали огромное число исследований, направленных на развитие метода газовой хроматографии.
За короткое время были усовершенствованы конструкции систем ввода проб, созданы чувствительные детекторы. Метод газовой хроматографии - первый из хроматографических методов, получивших инструментальное обеспечение. Начиная с 70-х годов происходит бурное развитие жидкостной хроматографии. К настоящему времени разработаны теория хроматографического процесса и множество хроматографических методов анализа.
Среди разнообразных методов анализа хроматография отличается самой высокой степенью информативности благодаря одновременной реализации функций разделения, идентификации и определения. Кроме того, метод используется и для концентрирования. Хроматографический метод анализа универсален и применим к разнообразным объектам исследования (нефть, лекарственные препараты, вещества растительного и животного происхождения, биологические жидкости, пищевые продукты и др.). Хроматография отличается высокой избирательностью и низким пределом обнаружения. Эффективность метода повышается при его сочетании с другими методами анализа, автоматизацией и компьютеризацией процесса разделения, обнаружения и количественного определения.
2. Классификация методов хроматографии
Различные методы хроматографии можно классифицировать по агрегатному состоянию фаз, механизму разделения, аппаратурному оформлению процесса (по форме) и по способу перемещения подвижной фазы и хроматографируемой смеси.
По агрегатному состоянию фаз различаютжидкостную и газовую хроматографию.
Разделение веществ протекает по разному механизму, в зависимости от природы сорбента и веществ анализируемой смеси.
По механизму взаимодействия вещества и сорбента различают сорбционные методы, основанные на законах распределения (адсорбционная, распределительная, ионообменная хроматография и др.), гельфильтрационные (проникающая хроматография), основанные на различии в размерах молекул разделяемых веществ. На практике часто реализуются одновременно несколько механизмов разделения.
По технике выполнения хроматографию подразделяют на колоночную, когда разделение веществ проводится в специальных колонках, и плоскостную: тонкослойную и бумажную. В тонкослойной хроматографии разделение проводится в тонком слое сорбента, в бумажной - на специальной бумаге.
В зависимости от агрегатного состояния фаз, механизма взаимодействия и оформления различают основные виды хроматографии, которые приведены в табл. 1.
Таблица 1
| Вид хроматографии | Подвижная фаза | Неподвижная фаза | Форма | Механизм разделения |
Газовая: Газоадсорбционная Газожидкостная | Газ Газ | твердая жидкость | колонка колонка | Адсорбционный Распределительный |
Жидкостная: Твердожидкостная Жидкость-жидкостная Ионообменная Тонкослойная (т/ж) Тонкослойная (ж/ж) Бумажная Гельпроникающая (молекулярно-ситовая) | жидкость жидкость жидкость жидкость жидкость жидкость Жидкость | твердая жидкость твердая твердая жидкость жидкость жидкость | колонка колонка колонка тонкий слой тонкий слой лист бумаги колонка | Адсорбционный Распределительный Ионный обмен Адсорбционный Распределительный Распределительный по размерам молекул |
В соответствии с режимом ввода пробы в хроматографическую систему различаютфронтальную, элюентную и вытеснительную хроматографию. Если растворенную смесь непрерывно вводить в хроматографическую колонку, то в чистом виде можно выделить только одно, наиболее слабо сорбирующееся вещество. Все остальные выйдут из колонки в виде смеси. Этот метод называют фронтальным. В элюентном режиме через колонку пропускают подвижную фазу (элюент), вводят пробу, затем снова пропускают подвижную фазу (ПФ). В процессе движения по колонке компоненты смеси разделяются на зоны. Эти зоны поочередно выходят из колонки, разделенные зонами чистого растворителя.
В вытеснительном методе после введения пробы и предварительного разделения слабоактивным элюентом состав элюента меняется таким образом, что он взаимодействует с неподвижной фазой (НФ) каждого из компонентов анализируемой смеси. Вследствие этого новый элюент вытесняет компоненты, которые выходят из колонки в порядке возрастания взаимодействия с НФ. В этом методе не достигается достаточно полное разделение из-за частичного перекрывания зон.
Наибольшее распространение получил элюентный режим хроматографирования, позволяющий получать в чистом виде все компоненты пробы.
В жидкостной хроматографии применяют изократический и градиентный режим подачи элюента. В изократическом режиме состав элюента в течение анализа не изменяется, а в градиентном режиме состав элюента меняется по определенной программе.
Рассмотрим особенности отдельных наиболее широко применяемых видов хроматографии.
3. Жидкостно-адсорбционная хроматография на колонке
Разделение смеси веществ в адсорбционной колонке происходит в результате различия их в сорбируемости на данном адсорбенте (в соответствии с законом адсорбционного замещения, установленного М.С.Цветом).
Адсорбентами являются пористые тела с сильно развитой внутренней поверхностью, удерживающие жидкости с помощью межмолекулярных и поверхностных явлений. Это могут быть полярные и неполярные неорганические и органические соединения. К полярным адсорбентам относятся силикагель (высушенная желатинообразная двуокись кремния), оксид алюминия, карбонат кальция, целлюлоза, крахмал и др. Неполярные сорбенты - активированный уголь, порошок резины и множество других, полученных синтетическим путем.
К адсорбентам предъявляют следующие требования:
- они не должны вступать в химические реакции с подвижной фазой и разделяемыми веществами;
- должны обладать механической прочностью;
- зерна адсорбента должны быть одинаковой степени дисперсности.
При выборе условий для хроматографического процесса учитывают свойства адсорбента и адсорбируемых веществ.
В классическом варианте жидкостной колоночной хроматографии (ЖКХ) через хроматографическую колонку, представляющую собой стеклянную трубку диаметром 0,5 - 5 см и длиной 20 - 100 см, заполненную сорбентом (НФ), пропускают элюент (ПФ). Элюент движется под воздействием силы тяжести. Скорость его движения можно регулировать имеющимся внизу колонки краном. Анализируемую смесь помещают в верхнюю часть колонки. По мере продвижения пробы по колонке происходит разделение компонентов. Через определенные промежутки времени отбирают фракции выделившегося из колонки элюента, который анализируют каким-либо методом, позволяющим измерять концентрации определяемых веществ.
Колоночная адсорбционная хроматография в настоящее время применяется, главным образом не как самостоятельный метод анализа, а как способ предварительного (иногда и конечного) разделения сложных смесей на более простые, т.е. для подготовки к анализу другими методами (в том числе и хроматографическими). Например, на колонке с окисью алюминия разделяют смесь токоферолов, пропускают элюент и собирают фракцию a-токоферола для последующего определения фотометрическим методом.
3.1. Высокоэффективная жидкостная хроматография
Хроматографическое разделение смеси на колонке вследствие медленного продвижения ПФ занимает много времени. Для ускорения процесса хроматографирование проводят под давлением. Этот метод называют высокоэффективной жидкостной хроматографией (ВЖХ)
Модернизация аппаратуры, применяемой в классической жидкостной колоночной хроматографии, сделала ее одним из перспективных и современных методов анализа. Высокоэффективная жидкостная хроматография является удобным способом разделения, препаративного выделения и проведения качественного и количественного анализа нелетучих термолабильных соединений как с малой, так с большой молекулярной массой.
В зависимости от типа применяемого сорбента в данном методе используют 2 варианта хроматографирования: на полярном сорбенте с использованием неполярного элюента (вариант прямой фазы) и на неполярном сорбенте с использованием полярного элюента - так называемая обращенно-фазовая высокоэффективная жидкостная хроматография (ОфВЖХ).
При переходе элюента к элюенту равновесие в условиях ОфВЖХ устанавливается во много раз быстрее, чем в условиях полярных сорбентов и неводных ПФ. Вследствие этого, а также удобства работы с водными и водно-спиртовыми элюентами, ОфВЖХ получила в настоящее время большую популярность. Большинство анализов при помощи ВЖХ проводят именно этим методом.
Аппаратура для ВЖХ
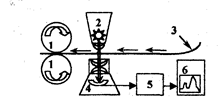
Комплект современного оборудования для ВЖХ, как правило, состоит из двух насосов 3, 4 (рис.3.1.1), управляемых микропроцессором 5, и по дающих элюент по определенной программе. Насосы создают давление до 40 МПа. Проба вводится через специальное устройство (инжектор) 7 непосредственно в поток элюента. После прохождения через хроматографическую колонку 8 вещества детектируются высокочувствительным проточным детектором 9, сигнал которого регистрируется и обрабатывается микро-ЭВМ 11. При необходимости, в момент выхода пика автоматически отбираются фракции.
Рис. 3.1.1. Схема современного жидкостного хроматографа
1,2 - сосуды с элюентами; 3, 4 - насосы; 5 контроллер;
6 - смесительная камера; 7 - инжектор; 8 - колонка; 9 - детектор;
10 - регистратор; 11 - блок автоматической обработки результатов анализа; 12 — коллектор фракций; 13- термостат
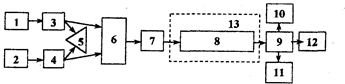 Колонки для ВЖХ выполняют из нержавеющей стали с внутренним диаметром 2-6 мм и длиной 10-25 см. Колонки заполняют сорбентом (НФ). В качестве НФ используются силикагель, оксид алюминия или модифицированные сорбенты. Модифицируют обычно силикагель, внедряя химическим путем в его поверхность различные функциональные группы.
Колонки для ВЖХ выполняют из нержавеющей стали с внутренним диаметром 2-6 мм и длиной 10-25 см. Колонки заполняют сорбентом (НФ). В качестве НФ используются силикагель, оксид алюминия или модифицированные сорбенты. Модифицируют обычно силикагель, внедряя химическим путем в его поверхность различные функциональные группы.
Детекторы. Регистрация выхода из колонки отдельного компонента производится с помощью детектора. Для регистрации можно использовать изменение любого аналитического сигнала, идущего от подвижной фазы и связанного с природой и количеством компонента смеси. В жидкостной хроматографии используют такие аналитические сигналы, как светопоглощение или светоиспускание выходящего раствора (фотометрические и флуориметрические детекторы), показатель преломления (рефрактометрические детекторы), потенциал и электрическая проводимость (электрохимические детекторы) и др.
Непрерывно детектируемый сигнал регистрируется самописцем. Хроматограмма представляет собой зафиксированную на ленте самописца последовательность сигналов детектора, вырабатываемых при выходе из колонки отдельных компонентов смеси. В случае разделения смеси на внешней хроматограмме видны отдельные пики. Положение пика на хроматограмме используют для целей идентификации вещества, высоту или площадь пика - для целей количественного определения.
Качественный анализ
Рис.3.1.2. Параметры хроматограммы
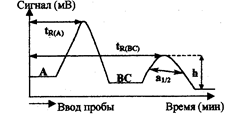 Важнейшие характеристики хроматограммы - время удерживания trи связанный с ней удерживаемый объем — отражают природу веществ, их способность к сорбции на материале неподвижной фазы и, следовательно, при постоянстве условий хроматографирования являются средством идентификации вещества. Для данной колонки с определенными скоростью потока и температурой время удерживания каждого соединения постоянно (рис), где tR(a) - время удерживания компонента А анализируемой смеси с момента ввода в колонку до появления на выходе из колонки максимума пика, tR(BC) - время удерживания внутреннего стандарта (первоначально отсутствующее в анализируемой смеси вещество), h - высота пика (мм), a1/2 — ширина пика на половине его высоты, мм.
Важнейшие характеристики хроматограммы - время удерживания trи связанный с ней удерживаемый объем — отражают природу веществ, их способность к сорбции на материале неподвижной фазы и, следовательно, при постоянстве условий хроматографирования являются средством идентификации вещества. Для данной колонки с определенными скоростью потока и температурой время удерживания каждого соединения постоянно (рис), где tR(a) - время удерживания компонента А анализируемой смеси с момента ввода в колонку до появления на выходе из колонки максимума пика, tR(BC) - время удерживания внутреннего стандарта (первоначально отсутствующее в анализируемой смеси вещество), h - высота пика (мм), a1/2 — ширина пика на половине его высоты, мм.
Для идентификации вещества по хроматограмме обычно используют стандартные образцы или чистые вещества. Сравнивают время удерживания неизвестного компонента tRx с временем удерживания tRCT известных веществ. Но более надежна идентификация по измерению относительного времени удерживания
tR(A)
tR(отн)= ____ (3.1.1).
tR(BC)
При этом в колонку сначала вводят известное вещество (внутренний стандарт) и измеряют время его удерживания tR(BC), затем хроматографически разделяют (хроматографируют) исследуемую смесь, в которую предварительно добавляют внутренний стандарт. Относительное время удерживания определяют по формуле (3.1.1).
Количественный анализ
В основе этого анализа лежит зависимость высоты пика h или его площади S от количества вещества. Для узких пиков предпочтительнее измерение h, для широких размытых - S. Площадь пика измеряют разными способами: умножением высоты пика (h) на его ширину (а1/2), измеренную на половине его высоты (рис 3.2.3); планиметрированием; с помощью интегратора. Электрическими или электронными интеграторами снабжены современные хроматографы.
Для определения содержания веществ в пробе используют в основном три метода: метод абсолютной градуировки, метод внутренней нормализации и метод внутреннего стандарта.
Метод абсолютной градуировки основан на предварительном определении зависимости между количеством введенного вещества и площадью или высотой пика на хроматограмме. В хроматограмму вводят известное количество градуировочной смеси и определяют площади или высота полученных пиков. Строят график зависимости площади или высоты пика от количества введенного вещества. Анализируют исследуемый образец, измеряют площадь или высоту пика определяемого компонента и на основании градировочного графика рассчитывают его количество.
Метод внутренней нормализации основан на приведении к 100% суммы площадей всех пиков на хроматограмме. Расчет массовой доли в % одного компонента проводят по формуле
KASA
w(a)% =________________ , (3.1.2)
KASa+KbSb+...K2Si
где К - поправочные коэффициенты, sa, sb, Si - площади пиков компонентов смеси.
Этот метод дает информацию только об относительном содержании компонента в смеси, но не позволяет определить его абсолютную величину.
Метод внутреннего стандарта основан на сравнении выбранного параметра пика анализируемого вещества с тем же параметром стандартного вещества, введенного в пробу в известном количестве. В исследуемую пробу вводят известное количество такого стандартного вещества, пик которого достаточно хорошо отделяется от пиков компонентов исследуемой смеси (рис. 3.2.3). Проводят анализ пробы с внутренним стандартом и рассчитывают количество определяемого вещества по формуле
k(a)h(a)
g(а)= ___________g(BC) (3.1.3)
K(BC)h(BC)
где g(A) - количество определяемого компонента А; h(A) - высота пика компонента A; g(BC)- количество внутреннего стандарта; h(BC) - высота пика внутреннего стандарта; к(A) и k(BC)- поправочные коэффициенты.
В последних двух методах требуется введение поправочных коэффициентов, характеризующих чувствительность используемых детекторов к анализируемым веществам. Для разных типов детекторов и разных веществ коэффициент чувствительности определяется экспериментально.
В жидкостной адсорбционной хроматографии используется также анализ фракций растворов, собранных в момент выхода вещества из колонки. Анализ может быть проведен различными физико-химическими методами.
Жидкостную адсорбционную хроматографию применяют в первую очередь для разделения органических веществ. Этим методом весьма успешно изучают состав нефти, углеводородов, эффективно разделяют - транс- и цис- изомеры, алкалоиды и др. С помощью ВЖХ можно определять красители, органические кислоты, аминокислоты, сахара, примеси пестицидов и гербицидов, лекарственных веществ и других загрязнителей в пищевых продуктах.
3.2. Ионообменная хроматография
Ионообменная хроматография (ИХ) является разновидностью жидкостной хроматографии и в аппаратурном оформлении ничем не отличается от других видов жидкостной колоночной хроматографии. В основе ионообменной хроматографии лежит процесс обмена между ионами анализируемого раствора (ПФ) и подвижными ионами того же знака ионообменника (НФ).
В качестве ионообменников или ионитов обычно используют синтетические полимерные вещества, называемые ионообменными смолами. Они состоят из матрицы (R) и активных групп, содержащих подвижные ионы. В зависимости от знака обмениваемых ионов различают катиониты и аниониты. Катиониты содержат кислотные группы различной силы, такие как сульфогруппы, карбоксильные, оксифенильные. Аниониты имеют в своем составе основные группы, например алифатические или ароматические аминогруппы различной степени замещенности (вплоть до четвертичных).
Иониты могут находиться в Н-форме и ОН - форме, а также в солевой форме. В Н-форме катиониты и ОН- форме аниониты содержат способные к обмену ионы водорода и гидроксила соответственно, в солевых формах ионы водорода заменены катионами металла, анионы гидроксила - анионами кислот.
В зависимости от силы кислотных и основных групп в ионитах различают сильнокислотные (R-SOзН) и слабокислотные (R-СООН) катиониты; сильноосновные (R-N(СНз)зОН) и слабоосновные (R-NНзОН).
Сильнокислотные и сильноосновные иониты способны к ионному обмену в широком диапазоне рН.
Процесс ионного обмена протекает стехиометрично. Например:
R-SO3H+Na+=RSO3Na+H+
R(NНз)зОН+Сl-=R(NНз)зСl+ОН-
Это ионообменное равновесие характеризуется константой ионного обмена:
(H+)(RSO3Na) (OH-)(RN(CH3)3Cl
KH+/Na+=______________; KOH-/Cl-= _________________
(Na+)(RSO3H) (Cl-)(RN(CN3)3OH)
На основании констант ионного обмена построены ряды сродства ионов к данному иониту, позволяющие предвидеть возможности ионообменных разделений.
В зависимости от сродства к фиксированным ионам неподвижной фазы разделяемые ионы перемещаются вдоль хроматографической колонки с различными скоростями; чем выше сродство, тем больше объем удерживания компонента. При разделении органических кислот и оснований важную роль играет степень их диссоциации.
Для двух веществ, имеющих разные константы обмене, рассчитывают фактор разделения или коэффициент распределения, который характеризует селективность ионита
KA
fa/b= ___ , (3.2.1)
KB
где fa/b - фактор разделения; KA; KB - константы ионного обмена веществ А и В. Чем больше фактор разделения, тем сильнее ионит удерживает вещество А.
Например, константы ионного обмена солей железа (III) и кобальта (II) на сильнокислотном катионите марки КУ-2 составляют 3726 и 286 соответственно.
3726
Тогда согласно формуле 7.2.1 получим: FFe3/Co2+ = ____=13.
286
Таким образом, можно сделать вывод, что катионит КУ-2 более селективен к ионам железа (III).
Важной количественной характеристикой ионитов является их обменная емкость. Полная обменная емкость определяется количеством эквивалентов ионов, обмениваемых одним граммом сухого ионита. Чем больше обменная емкость, тем большую пробу можно ввести в колонку с ионитом.
При подготовке ионитов к работе их переводят в соответствующую форму. Так, для перевода катионита в Н-форму через колонку с набухшим ионитом пропускают раствор сильной кислоты, избыток которой отмывают водой. Затем медленно пропускают раствор смеси ионов. Каждый катион задерживается на ионите согласно своей сорбируемости. Далее пропускают подходящий элюент. Например, катионы щелочных металлов легко элюируются 0,1 М HCl. При этом ионы водорода обмениваются на сорбированные катионы, которые вместе с раствором выходят из колонки в соответствии с константами ионного обмена. На выходе из колонки фракции собирают в отдельные сосуды и определяют содержание любым подходящим методом.
Иониты применяются для деионизации (обессоливания) воды, очистки сахарных сиропов от минеральных солей; в препаративной химии - для концентрирования растворов; для определения ионов железа (III), меди и свинца в вине; кальция и магния в молоке; различных металлов в биологических жидкостях. Кроме того, ионный обмен используют для перевода ионов в форму, удобную для количественного определения. Например, поваренную соль в рассоле можно определить, пропустив пробу через колонку с катионитом, и выделившуюся в эквивалентном количестве кислоту оттитровать щелочью:
R-SOзН+NaCI=R-SOзNa+НСl.
Ионообменную хроматографию применяют для разделения фенолов, карбоновых кислот, аминосахаров, пуриновых, пиримидиновых и других оснований. Часто иониты используют для предварительного разделения сложных смесей на менее сложные. На ионном обмене основано получение ионитного молока для детского питания. Ионный обмен используют для очистки натуральных соков от ионов тяжелых металлов. Ионообменные смолы применяют для получения ионообменных мембран.
3.3. Тонкослойная хроматография
Тонкослойная хроматография (ТСХ) является одним из наиболее простых и эффективных экспресс-методов разделения и анализа веществ в пищевых продуктах, биологических жидкостях и других объектах, не требующих сложного оборудования. В то же время метод обладает высокой избирательностью и чувствительностью (низким пределом обнаружения). Этим методом можно определить 10-20 мкг вещества с точностью до 5-7%.
В зависимости от природы НФ тонкослойная хроматография может быть адсорбционной и распределительной. Наиболее широко применим в ТСХ первый вариант разделения.
Неподвижная твердая фаза (оксид алюминия, силикагель и др.) тонким слоем наносится на стеклянную, металлическую (алюминиевая фольга) или пластмассовую пластинку, закрепляется слой с помощью крахмала или гипса (иногда используют пластинки с незакрепленным слоем). Для хроматографирования могут использоваться готовые пластинки, выпускаемые промышленностью, размером 5х15 или 20х20 см.
На расстоянии 2 см от края пластинки на стартовую линию с помощью микропипетки или микрошприца наносят пробы анализируемого раствора (диаметр пятен 3-5 мм). После испарения растворителя край пластинки помещают в стеклянную камеру, на дно которой налит растворитель (ПФ) в количестве, достаточном для образования слоя глубиной 0,5 см. Камеру закрывают крышкой.
Выбор растворителя (ПФ) зависит от природы сорбента и свойств анализируемых соединений. Например, разделение хлорорганических пестицидов на пластинке с силикагелем проводят в среде гексана. Часто применяют смеси растворителей из двух или трех компонентов. Так, при хроматографировании аминокислот используют смесь Н-бутанола с уксусной кислотой и водой, при анализе неорганических ионов - водные буферные растворы, создающие постоянное значение рН.
При хроматографировании растворитель движется снизу вверх (восходящий вариант) вдоль слоя сорбента и с разной скоростью переносит компоненты смеси, что приводит к их пространственному разделению. После окончания хроматографического процесса пластинку вынимают из камеры, отмечают линию фронта растворителя (обычно около 10 см) и высушивают.
Если компоненты смеси окрашены, то они четко видны на пластине после разделения. Неокрашенные соединения обнаруживают различными способами. Если пластину поместить в камеру с парами йода, то четко проявляются коричневые пятна для органических соединений с непредельными связями. Хроматограмму можно проявить, опрыскивая ее каким-либо реагентом, дающим с компонентами пробы окрашенные соединения. В состав нанесенного слоя в готовые пластины часто вводят люминофор. При облучении такой пластины ультрафиолетовым (УФ) светом она флуоресцирует, а разделенные компоненты пробы видны в виде темных пятен. Вещества, имеющие собственную флуоресценцию, также обнаруживают в УФ - свете (например, пестициды).
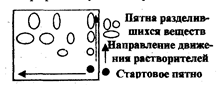
Идентификацию веществ на хроматограмме осуществляют по характеру окраски пятен, параметру удерживания Rf и с помощью стандартных веществ (свидетелей).
Величина Rf рассчитывается из экспериментальных данных по уравнению
l
Rf=__ , (3.3.1)
L
где l - расстояние от стартовойлинии до центра пятна, L - расстояние, пройденное за это же время растворителем (рис. 3.3.1).
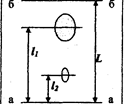 Рис. 3.3.1. Хроматограмма двухкомпонентной смеси
Рис. 3.3.1. Хроматограмма двухкомпонентной смеси
а - а: линия старта, в - в : линия фронта растворителя
При стандартных условиях величина Rf является постоянной величиной, характерной для данного соединения. Но практика показывает, насколько трудно создавать постоянство всех факторов, от которых зависит воспроизводимость значений Rf. На величину Rf влияет качество и активность сорбента, его влажность, толщина слоя, качество растворителей и другие факторы, не всегда поддающиеся достаточному контролю.
Рис. 3.3.2. Хроматограмма жира. I - полимеризованные и сильнополярные жиры; II - фосфолипиды, III – триглицериды 1 - говяжье мясо; 2 - свинина; 3 - свинина с 29% печени; 4 - свинина с 4% печени; 5 - свинина с 50% печени; 6 - свиная печень
Поэтому наряду с величиной Rf идентификацию проводят по “свидетелю”. Стандартное вещество (свидетель), наличие которого предполагают в анализируемой смеси, наносят на линию стандарта рядом с исследуемой пробой. Таким образом, стандартное вещество хроматографируется в тех же условиях. После хроматографирования и детекции пятен сравнивают величины Rf определяемого вещества и “свидетеля”.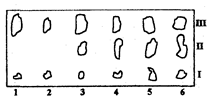
Качественный анализ после разделения компонентов смеси методом ТСХ часто используют для определения состава пищевых продуктов. Так, на рис. 3.3.2 представлена хроматограмма жира, выделенного из мясного фарша различного состава. Хроматографирование проводили на пластинках с силикагелем в системе гександиэтиловый эфир (в соотношении 3:1), пятна детектировали 10% раствором фосфорно-молибденовой кислоты, идентифицировали по голубому цвету зон на желтом фоне пластинки. Как видно из хроматограммы, при данных условиях произошло разделение фосфолипидов и триглицеридов. По характерному составу компонентов мяса и печени можно сделать вывод о натуральности мясного фарша в пробах 1-2, и добавках к нему печени в пробах 3-5.
Количественное определение в ТХС может быть проведено непосредственно на пластинке, иди после удаления веществ с пластинки. При непосредственном определении на пластинке измеряют тем или иным способом площадь пятна (например, с помощью миллиметровой кальки) и по заранее построенному градуировочному графику находят количество вещества. Зависимость между массой вещества q и площадью S на хроматограммах носит нелинейный характер и является логарифмической:
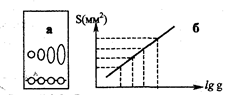 S=a lg q + в, (3.3.2)
S=a lg q + в, (3.3.2)
где а и в эмпирические константы. Эта зависимость линейна для количеств вещества от 1 до 80-100 мкг.
Рис. 3.3.3. Зависимость площади пятен на хроматограмме от количества вещества: а - хроматограмма, б – калибровочный график
Для построения градуировочного графика на пластинку наносят растворы, содержащие разные количества стандартного вещества, хроматографируют, проявляют зоны и измеряют их площади (рис. 3.3.3).
Более точен денситометрический метод определения веществ на хроматограммах (ошибка - 1-2%). В методе денситометрии производят измерение оптического поглощения проявленной хроматограммы сканирующим лучом в проходящем или отраженном свете на специальных приборах-денситометрах (рис.3.3.4.).
Рис. 3.3.4. Схема денситометра. 1 – протяжный механизм; 2 – источник света; 3 – хроматограмма, 4 – фотоэлектрический преобразователь, 5 – усилитель, 6 – самописец.
 На денситограмме получают пики, площадь которых пропорциональна содержанию вещества в пятне. Построив с помощью стандартов калибровочный график, измеряют площадь пика компонента и по графику определяют его массу в пробе. Получают развитие также спектрофотоденситометрическое и флуориметрическое определение веществ на хроматограммах.
На денситограмме получают пики, площадь которых пропорциональна содержанию вещества в пятне. Построив с помощью стандартов калибровочный график, измеряют площадь пика компонента и по графику определяют его массу в пробе. Получают развитие также спектрофотоденситометрическое и флуориметрическое определение веществ на хроматограммах.
В первом случае используют специальные спектрофотоденситометры, измеряющие поглощение вещества в монохроматическом свете, во втором измеряют флюоресценцию пятна при облучении хроматограммы УФ светом. Широкое распространение получил способ экстрагирования компонентов из зон подходящим растворителем. При применении этого способа на хроматограмму наносят стандартный раствор и раствор пробы. После получения хроматограммы производят ее обработку, детектируя зону стандарта, вырезают часть хроматограммы с зоной компонента пробы и производят его экстрагирование подходящим растворителем. Полученный раствор анализируют инструментальным методом, имеющим высокую чувствительность. Чаще всего применяют спектрофотометрические и фотоколориметрические методы. Если вещество не имеет цвета или не обладает поглощением в УФ-области, с экстрактом проводят фотометрическую реакцию, позволяющую получить интенсивно поглощающее производное вещества.
Тонкослойная хроматография находит применение при исследовании некоторых видов пищевых продуктов на безопасность. Например, для определения токсинов (афлатоксинов, микотоксинов, патулина и др.) в арахисе, в зерновых, овощах, фруктах, напитках; для определения пестицидов (ДДТ и др.) в растительных и животных продуктах, определения гистамина как показателя порчи рыбы. Кроме того, ТСХ часто сочетают с газовой хроматографией, электрофорезом и другими методами.
3.4. Хроматография на бумаге
По механизму разделения различают распределительную, адсорбционную, осадочную и другие виды бумажной хроматографии (БХ). В распределительной жидкость-жидкостной хроматографии бумага, приготовленная из специальных сортов хлопка, выполняет роль носителя неподвижной жидкой фазы (НФ), в качестве которой часто выступает вода, адсорбированная парами бумаги. В таком случае гидрофильная бумага используется для нормально-фазовой хроматографии.
Растворителями (ПФ) являются спирты (метанол, этанол, н-пропанол, бутанол), простые эфиры (этиловый, метиловый), кетоны (ацетон, ацетил-ацетон), эфиры органических кислот (метилацетат, этилацетат), пиридин, хлороформ. Чаще используются смеси растворителей. Так, для разделения неорганических неполярных веществ употребляют системы:
- ацетон: НCl: Н2О (в различных соотношениях);
- Н-бутанол, насыщенный НСl (различной концентрации);
- Н-бутанол: 0,1М НNОз - ацетилацетон.
Для разделения некоторых органических веществ используют метод обращенных фаз. В этом методе для придания бумаге гидрофобного характера ее импрегнируют (пропитывают) нафталином, парафином, раствором каучука, силиконом и др. Такая бумага служит носителем для неполярных растворителей в качестве НФ. В качестве ПФ применяют смеси кислот с низшими спиртами.
 Обращеннофазовая бумажная хроматография используется, например, для разделения и идентификации полинасыщенных жирных кислот при изучении состава липидов, выделенных из животных тканей. Бумагу пропитывают 5% раствором силикона, в качестве ПФ используют 85% раствор уксусной кислоты.
Обращеннофазовая бумажная хроматография используется, например, для разделения и идентификации полинасыщенных жирных кислот при изучении состава липидов, выделенных из животных тканей. Бумагу пропитывают 5% раствором силикона, в качестве ПФ используют 85% раствор уксусной кислоты.
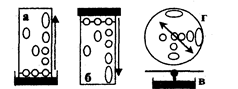
Рис.3.4.1. Виды бумажной хроматографии
Разделение веществ в распределительной БХ осуществляется благодаря различию в скоростях движения компонентов при многократном повторении актов экстракции и сорбции. Скорость перемещения компонентов зависит от их коэффициентов распределения (как и в методе экстракции).
По направлению движения элюента (ПФ) различают восходящую, нисходящую и радиальную (круговую) хроматографию.
Если элюент движется по бумаге вверх, метод называют восходящей (а) бумажной хроматографией; при его движении сверху вниз - нисходящей (б) бумажной хроматографией. Очень быстро можно осуществить хроматографический анализ методом радиальной (в) бумажной хроматографии, в котором используется бумажный круг (г) с фитилем, опущенным в элюент. (рис. 3.4.1)
 Иногда при сложном составе пробы не удается разделить ее компоненты с помощью одного растворителя. Тогда применяют двумерную хроматографию. В угол квадратного листа хроматографической бумаги наносят хроматографической бумаги наносят раствор пробы и хроматографируют сначала в одном элюенте, затем, повернув хроматограмму на 90, - в другом. Первый элюент производит предварительное разделение компонентов пробы, второй окончательное (рис.3.4.2).
Иногда при сложном составе пробы не удается разделить ее компоненты с помощью одного растворителя. Тогда применяют двумерную хроматографию. В угол квадратного листа хроматографической бумаги наносят хроматографической бумаги наносят раствор пробы и хроматографируют сначала в одном элюенте, затем, повернув хроматограмму на 90, - в другом. Первый элюент производит предварительное разделение компонентов пробы, второй окончательное (рис.3.4.2).
Рис.3.4.2. Двухмерная хроматография
Для проведе